Способ получения производных 2-тиенилоксиуксусной кислоты или их фармацевтических приемлемых солей
Номер патента: 1784
Опубликовано: 30.09.1997
Авторы: Франц Ровенсцки, Дитер Биндер, Хуберт Петер Фербер




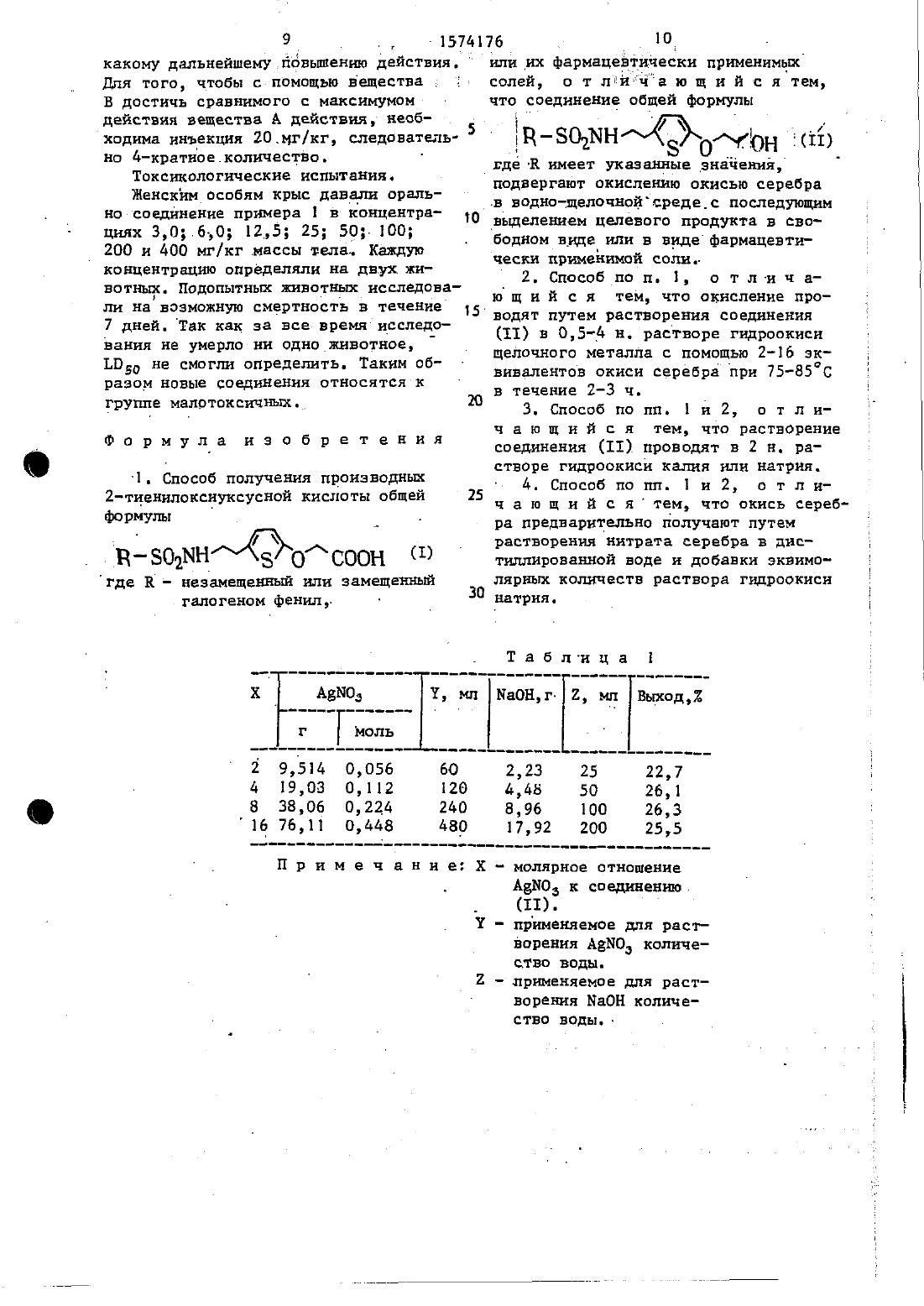

Текст
тровывают и промывают многократно дистиллированной водой.9,3 г (0,028 мольУашща Ы- 2-12(5-(2-окси)этокси)ттиеннл-этил- 5 оензолсульфокиспоты (1) растворах-от в 90 мп водного раствора гидроокисинатрия, добавляют еще влажную окись,серебра и при механическом перемешивании напревают до 80 С. Спустя 3 ч ю при этой температуре охлаждают и суспензии фильтруют через НЗЪО Прозрачный раствор гидроокиси натрия подкисляют с помощью примерно б мл,концентрированной солянойкислоть 1 и 15 экстрагируют трижды по 100 мл эфира. Эфирные фазы встряхивают триэкды по 100 мл с насьппенъгьш раствором бикарбоната натрия, промывают их 50 мл эфира и подкнсляют концентрированной 20соляной .кислотой. Воднуто фазузкстра тируют дважды с помощью 15 О шт эфира, объединенные эфирные фазы сушат над сульфатом натрия, опфильтровъгва ют и выпаривают, Кристаллический 23 .осадок отстаивают с 30 мл диизопропилового эфира и отфильтровывают.Исходный продукт можно получать следующих образом 50к 1600 мл абсолютного этиленглн- коля добавляют 321,7, мл 5,51 Цраствора метнлата натрня(1,751 моль) Реакци онную смесь нагревают. и. образующнйсэт 55 метанол отгоняют путем пропускания азота через обратный холодильник до тех пор, пока температура в шпане-й части не повысится до 130. По окон чании удаления метанола добавляют187,5 1 (115 моль) 2-бромтиофена,- 55,5 тдтонко измельченной окиси меди и 5,0 г иодида натрия, аппаратуру еще короткое-время промывают азотом,закрывают баллон и перемешивают 175 ч при 80 С. Затем реакционную смесь охлаждают и отсасывают через НУЕЬО. Фильтрат разбавляют 800 мл воды и слегка цодкислятот концентрированной соляной кислотой. .экстрагируют четырераза по 1100 мл метиленхлоридом (в целом 1600-1111). Объединенные органические, фазы встряхивают один раз с 200 мл воды, сушат над Асульфатом натрия, отфнпьтровывают и ВЬШарНБЗЮТи Остаток перегоняют.30 г (0,208 моль) 2-(2 т-тиенилок отд-этанола помещают в 300 мл абсолютного тетрагидрофурана ирастворяют в этом растворе 50 мг п-толуол суньфокислотьх. К раствору добавляют 18,37 г 03,218 молъ) ЗА-дигидропирана и перемешивают 8 ч. 1 .03,208 моль) -2,5 И раствора н-бутил у питиядз н-гексане так, чтобы тем пература не превышала -10 с. оставляют нагреваться до комнатной температуры и перемена-хвалит в течение часа. 11 Реакционную смесь онлазкдаютдо 10 С и в течение 30 мин прикапьшадот раствор 19,05 г (0104 моль) п-бензолсупьфонилазиридина в 100 мл абстлютного ТГФ при 1 О-15 С. Все нагревается до комнатной температуры и перемешивается еще 2 ч. р-Смесь выливают на 200, мл 2 н. водного раствора НС 1 и экстрагируют трижды по 120 мл метилешшорндом. Объединенные органические фазы сушат Над СУЛЪФатом натрия, отфильтровывают и вьшариваюъ. Остаток обрабатывают 300 мл абсолютного метанол-телемешагают с.2 мл ЗОХ-ной метанольной соляной кислоты и перемешивают 10 мин при комнатной темпер-атуре. После добавки 2 г карбонатанатрия вьшаривают а вакууме.Остаток распределяют между 250.мл 1 нводного раствора гидроксида5, 157 д натрия и 200 мл эфира, и эфирную фа эу дополнительно промывают один раз 50 мл 1 Н. раствора гидроксида натрил. Объединенные водные фазы 5промывают дважды по 10 мл эфиром,подкисляют примерно 25 мл концент рироваиной соляной кислоты и экстрагируют трехкратно, смотря по обстоятельствам, 150 мл-метиленхлорида. Ме- 10 тиленхлоридную фазусушатнадсУЛЬфН том натрий, смешивают с 3 г активного угля, отфильтровывают и выпаривач ют. Выход 33,3 г темноткрасного масла (978 от теории)ш которое используется непосредственно на самой блнайшей стадии. 1 .П р и м е р 2. Растворяют 1 г 52-(бензолсульфониламмно)этил-2 тиеннлоксиуксусной Кислоты (2,93 ммолЫ в 20 мл метанола И прикалывают 0,117 г Ыабн (2,9 З ммолъ), растворенных в 10 мл метанола, и перемешивают раствор в течение 1 ч при комнатной температуре. В заключение упаривают досуха, остаток промывают этилацетатом и диизопропиловым эфиром И сушат в вакууме при 5090.(0,02 В моль) амида 111-2- 2- (5-(2 гидрокси)гэтокси)-тиенил-Этилбенэолсульфоновой кислоты (11) в 90 мл 2 М.водной натриевой щелочи каждая,добавляют еще влажную окись серебра.и нагревают при перемешивании додальнейшаяобработка происходит как в.прнмере 1. Все данные сведены в табл. 1.115 г (0 О 88 моль) нитрата серебра растворяют В 90 мл дистиллированной воды и при перемешивании медленно прикалывают раствор 3,5 г-45 мл дистиллированной воды, Образо вавшуюся суспензию окиси серебра перемешивают еще 10 мин, осадок отфиль тровьшают и промывают многократно дистиллированной водой.дО г (0,01 моль) амидад-хлорЫ-2-2(5(2-окиси)этокси)ттиенил-7 этил бензолсулвфокислоты(111 растворяют в ДО мл 2 н. водного раствора гидроокиси натрия, добавляют еще влажную окись серебра и нагревают до 8 ОС при механическом перемешива.нии, СПУСТЯ 3,5 Ч при этой темпера туре охлаждают и суспензию отсасывают через НЪ 0 и дополнительно промывают 2 н. водным раствором гидрооки- си натрия Прозрачный раствор гидроокиси натрия подкисляют с помощью концентрированной соляной кислоты и экстрагируют триды по 80 мл эфиром.Эфирные фаэылпажды встряхивают с насыщенным раствором бикарбоната натрия, отбирая каждый раз по 50 мл,промывают один раз 5 Омл эфира и подкисляют концентрированной соляной кислотой. водную фазу экстрагируют дважды по 150 мл эфиром, объединенные эфирные фазы сушат над сульфатом натрия, отфильтровывают и выаривают. Сырой продукт перекристаллизуют из толуола. Вьшод 1,3 г бесцветных кристаллов (З 1,7 от.теории) т.пл 125127 С (толуол). 1температура не превьщала -15 С.43,8 трнплет ПЦСН 1) 29,9 (три-5 (15 от теории), т.пл. 8-87 С (бен плет ТЬ-Снд). 3 вол). . исходны продукт может быть полу-5 исследование.антитронботнческой 1самцов крыс И 1 вЬаг (ЭР) массой 200-300 г.наркотизируют с помощью пентобарбиталнатрии (60 мг/кг интраперитонеально).После этого животным 10 внутривенноинъекцируют вещество примера 1 (вещество А) даэоксибен гидроклорид д-2-(1 Н-нмидазол-1-ил)сульфокислоты. Добавляют 14,2 г этоксибензойной кислоты (вещество(0169 ноль) З,Ьдигидропирана и В Венолубрьшейки) соответственно. р перемешивают 8 чы Охлаждают до 209 С 15 высвобождают препарированием укрепи при перемешивании прикалывают - ляют в скобках на объективе микроско 66 мл (0,165 моль) 2,5 М раствора па н ополаскивают с постоянной сконбутиллитняв нгексане так, чтобы ростью 2,5 мл/мин физиологическим раствором хлорида натрия. Лазерный 20 луч когерентности СЕ 2 суперграфитноге ионного лазера (аргоновый лазер) в течение 30 мин после инъекции Испытуемого вещества направляют через интерференционно-коитрастный объектив 25 50 микроскопа Ьейфа .0 г 1 Ьор 1 ао с продолительиостью 1/30 с на венолу ИсЫодная энергия ниже объектива микроскопа составляет 0,18 Вт. Если после первого лазерного пуска не образует 3 о ся никакой тромбоцитный тромб или. если тромб по длине и ширине не соответствует диаметру сосуда, то проиаводятвторой запуск лазера, чтобы получить тромб, который по длине и 35 ширине соответствует диаметру сосуда. . Число лазерных пусков при этом явНОЙ КНСд 9 ТЫ И ПЕРЕМЕШИВЭЮТ 10 МИН ляется мерой антитромботической активпри комнатной температуре. Добавляют ности испытуемых веществ чем больше одну ложку карбоната натрия и метанол 40 число лазерных пусков при.одинаковом отгоняют. Остаток распределяют меж- диаметре сосуда, тем сильнее антиду 100.д 1 н. водного раствора тромботический эффект ГИДРОКСИДЗ натрий И 100 МЛ эфира И 1 В качестве контролей служат жиЭФНРНУЮ ФгЭУд 0 ППНИТеЛЬН 0 промывают нотные, которые не получили никаких ОДИН Раз 100 МЛ 1 Нь ВВСТБОРЭ ГИдР 0 45 испытуемых веществ. . окиси натрия, объединенные водные Тесты осуществляются по испытуемоФЗЗЫ ПРОНЫВЗШТ дВгЖды.по 50 мл эфиром, МУ веществу и концентрации на 5 жиподкисляют концентрированной соляной вотных, причем у животного повреждакислотой и экстрагируют трижды, емот- штся 3 сосуда диаметром 20-30.мм. РЯ ПО Обстддтедьствдмэ-100 МЛ МЕТИПеН 5 о Статистнческаяоценка осуществляетсяхлорида. Затем сушат надчсульфатом по тесту кгизка 1 и Ыа 111 э.и по КапеНаТрНН, смешивают С активным УГЛЕМ, ацшшеп тесту по Пцппг0 ТФНдЬТР 0 ВЫВ 3 ШТ И ВЫПаРИЕ 3 ЮТ- ПОЛУ Результаты опытов представлены в чают 10,15 г вязкого темного масла. табл. 2 .Этот сильно загрязненный сырой-про- Начало антнтромбического действия дукт фильтруютчерез силикагель 60 у вещества А находится при 1 мг/кг(180 г силикагеля, элюирующее средст массы тела и достигает максимума дейво, этилацетат/петролейный эфир). ствия при 5 мг/кгДальнейшее увелиВЫШОД 4,5 ГбЕСЦВеТНЫХ кристаллов чение концентрации не приводит ни к18,9 г (0137 моль) 2(2-тиеннлокси)-этанола помещают в 200-мл абсолютного тетрагицрофурана и растворяют там примерно 50 мг п-толуол Оставляыт медленно нагреваться до КОМНЭТЧЧ ТЕМПРаТУры и перемешивают далее 1 ч. 0Теперь при -50 РС прикалывают раствор 18 г (0,0 В 3 моль) Н(4 хлор беда 0 лсУлЬФонил)гезиридина в ОО мл абсолютного ТГФ. Реакционную смесь нагревают до комнатной температуры и перемешивают еще 30 миньРеакционную смесь выливают на-200 мл 2 и. водной НС 1 и экстрагируют триды по 250 мл.метиленхлоридом. Объединенные органические фазы сушат над сульфатом натрия, отфильтровывают и выпаривают. Остаток обрабатывают 100 мл абсолютного метанола, смеши 55Для того, чтобы с помощью вещества 1 В достичь сравнимого с максимумом действия вещества А действия, необходима инъекция 2 Омг/кг, следовательно 4-кратное.колиЧестВо.Женскимособям крыс давали орально соединение примера 1 в концентрацинк 30 б,0 25 25 50-100 200 и 400 мг/кг.массы тела Каждую концентрацию определяли на двух животныи. подопытных животных исследовали на возможную смертность в течение 7 дней. так как за все время исследования не умерло ни одно животное, Ьпдд не смогл определить. Таким образом новые соединения относятся к группе малртоисичны.1. Способ получения производны 2 тиенилокснуксусной кислоты общейгде к- неаамещенны или замещенный галогеном Фенил, ли- ц-м--нилинх фармацевтически применимыг солей, о т лн ча ю щ и й с я тем,что соединение общей формулыу 1 - 5 1 4 1 . ТЦ-ЗО-дЫН 8 ОАКЬН 111) где-Е имеет указанные значение, подвергают окислению окисью серебравыделенем целевого продукте в сво бодном виде или в видефармацевтически применимой солвг 2. Способ по п. 1, ю.щ и й с я15 водят путем растворения соединения(11) в 0,5 д н. растворе гидроокиси щелочного металла с помощью 2-16 эквивалентов окиси серебра при 75 т 85 С в течение 2-3 ч.3. Способ по пп. Е и 2, о т л ич а ю щ и й с я тем, что растворение соединения (11) проводят в 2 н. растворе гидроокиси каля или натрия. 25 4.Шюшбпопш 1 и 2, отлич а ю щ и й с я тем, что окись серебра предварительно получают путем растворения нитрата серебра в дистиллированной воде и добавки эквимо лярных количеств раствора гидроокиси натрия.Х Ш молярное отношение Авыоз к соединению.Т - применяемое для растворения А 3 Ы 0 д количество воды.2 применяемое для растворения НаОН количество воды.
МПК / Метки
МПК: A61K 31/38, C07D 333/26
Метки: способ, производных, или, 2-тиенилоксиуксусной, солей, кислоты, приемлемых, фармацевтических, получения
Код ссылки
<a href="https://by.patents.su/6-1784-sposob-polucheniya-proizvodnyh-2-tieniloksiuksusnojj-kisloty-ili-ih-farmacevticheskih-priemlemyh-solejj.html" rel="bookmark" title="База патентов Беларуси">Способ получения производных 2-тиенилоксиуксусной кислоты или их фармацевтических приемлемых солей</a>
Способ получения производных 6-фтор-1,4 дигидро-4-оксо-7-замещенной пиперазинилхинолин-3-карбоновой кислоты или их фармацевтически приемлемых солей
Номер патента: 605
Опубликовано: 30.06.1995
Авторы: Нобуо Огава, Эйити Косинака, Томио Сузуки, Ясуо Итох, Нориюки Яги, Хидео Като
МПК: A61K 31/495, C07D 401/02
Метки: кислоты, фармацевтически, производных, пиперазинилхинолин-3-карбоновой, приемлемых, 6-фтор-1,4, или, дигидро-4-оксо-7-замещенной, солей, способ, получения
Текст:
...игл, тдш. 261 2 в 2,5 с. о Вычислено, 2 С 54,74 Н 3,53 М 4,31. 7 99107131103 ч Найдено, С 34,64 н 3,47 Ы 1553 П р и м е р 5.- (К)с 1 гЭпш- д 6 , 8-дифтор 1 дг-дигидротР-(Згчметилпъновая кислота. а Смесь ДОП г 191 Ш 67,81 рИфторг 1 , дътдигидроттоксошнозшн-3 карбоногтУ войдкнслоты, 1,51) г (к)2 чметуш-. пиперазина,ы - 6,4 с, эта- нол),и 15 мл пиридина нагревают в течение 15 щи при дефлегмированни. После завершения реакшщ...
Способ получения замещенных аминодикислот, их рацематов или оптических изомеров, или их фармацевтических приемлемых солей

Номер патента: 479
Опубликовано: 30.03.1995
Авторы: Жорж Ремон, Мишель Винсен, Мишель Лоби
МПК: A61K 31/47, C07D 217/26, C07D 209/42...
Метки: фармацевтических, рацематов, аминодикислот, способ, изомеров, оптических, получения, или, замещенных, приемлемых, солей
Текст:
...ха. Остаточньт сложньт амноэфир 5 фазу подкислиют 4,4 мл 1 нНС 1. Обра(504 г) растворяют в 30 мл диметил- зовавшися осадок экстрагируют 2 раформамнда и этот-раствор добавляют за по 20 мл этилацетата, полученньй к перемешиваемому раствору 5 г раствор сушат над СаО 4, фильтруют н(0,02 б 4 моль) третбутилкарбонил- выпаривают. Полученный остаток пред(5)алаиина в 30 мл диметилформамит 10 ставляет собой искомы продукт,да, охлажденного до...
Способ получения сульфинильных производных гетероциклических соединений или их фармацевтически приемлемых солей

Номер патента: 77
Опубликовано: 30.09.1994
Авторы: Энтони Говард Инголл, Джон Льюис Сучитский, Дэвид Кокс
МПК: C07D 263/58, A61K 31/415, C07D 235/28...
Метки: получения, производных, сульфинильных, или, гетероциклических, солей, соединений, способ, приемлемых, фармацевтически
Текст:
...25 меру 1 а. 7 - 3(1 Н 2 Бензимидазолипсульфиннл метил)-ЫПдиметил 2 пиридинамин (е)г Продукт ц превращают в целевое соединение (т.пл. 12 д 126 С)по прн- за меру 1 Ь П р им е р 5. 2-(Н 2 Бензиидазолилсульфинилметил)дбенволамнн. Ы 2 дХлорметнлфенил 32 д,6 триметилбенволсульфонамнд (а). Ы(2 Гидроксиметилфенил)-24,6-триметилбенэопсулЬфонамид(40 Г) в сухом дихлорэтана (80 мл) обрабатывают тионипхлоридом 01,15 ил) ПРИ КОМ натной температуре...
Способ получения производных 1H-1,2,4-триазола или их фармакологически приемлемых кислотно-аддитивных солей

Номер патента: 333
Опубликовано: 30.12.1994
Авторы: Лео Баккс, Ян Херес, Жозеф Мостман
МПК: A01N 43/64, C07D 249/08
Метки: получения, приемлемых, 1h-1,2,4-триазола, производных, фармакологически, способ, или, кислотно-аддитивных, солей
Текст:
...охлаждают до комнатной температуры и продукт экстрагируют 765 вес.ч. бензола. Экстракт промывают 10-ным,раствором едкого натра, высушивают, фильтруют н отгоняют растворитель. Остаток дваждыП р и м е р 2. Согласно методике, описанной в примере 1 применяя эквивалентное количество соответствующего замещенного фенола вместо 4 клор-3-метнлфенола получают следующие промежуточные продукты 1 С 3-бромпропоксиЪ 4-хлор-2 метилбензол, т.кип. 115-11...
Способ получения правовращающих производных оксиизоиндолинила или их фармацевтически приемлемых солей

Номер патента: 539
Опубликовано: 30.06.1995
Авторы: Фабрицио Орци, Бруно Миорини, Джорджио Черони, Пьерлуиджи Гриджи, Джованни Карниель
МПК: C07D 209/46
Метки: правовращающих, или, фармацевтически, солей, производных, оксиизоиндолинила, приемлемых, способ, получения
Текст:
...кальция, КОТОРЫЙ после нагревания до 5 ОСдобавляют промывают водой при комнатной температ 2 о (цн 4)25(1 мл) и осадок фидьтруют ТУге. Маточные воды после ЦВНТРНФУГИ под вакуумом.К отфищьтрованному РаСТРования и промывки соединяют и добавд 55 вору добавляют при перемешивании 99. ляют гидроокись натрия 35 Ве (10,1 г) муравьиную кислоту до рН 3,5- 4. По Поставленная цель достигается тем,что соединение общей формулы (113Реакционную смесь нагревают...
Предыдущий патент: Способ получения сыра
Следующий патент: Способ получения лейкоцитарного интерферона
Случайный патент: Способ термовакуумной металлизации феррит-гранатов