Способ получения замещенных аминодикислот, их рацематов или оптических изомеров, или их фармацевтических приемлемых солей








Текст
Е - низшая алкильная группа с Ь 1-Ц атомами углерода, содерд жащая аминотруппуК атом водорода или алкильная группа с 1-4 атомам углеродеКЭ линейная ил разветвленная алкильная группа с 1-8 ато мами углеродамоно или ди-20 циклоалкил-алкильная группа или фенил-алнльнал грулпа содержащая в целом до 9 атомов глерода или замещенная алильнап группа формулыВТ - гпцроксильный нлп низший алкоксильный радикалы, ПОД вергают реакции восстановительного алкилирования С соедннением общей ФОРМУЛЫ 1115где К и На имеют указанные значения,С ВЫДЕЛЕНИЕ) ЦЕЛЕВОГО ПРОДУКТЕ ИЛИ со снятием в случае необходимости Ызашмтньш и эфирных групп гидрогенолизом или омылением. ПРИОРИТЕТ ПО ПВИЗНЗКЗМалкильная группа с 1-8 атома 4 МН углерода МОНО- ИЛИ ДНЦНК лоалкил-алкильнан группа или ФЕННТГЧЛКНЛЬНЭЯ группа, СО держащая в целом до 9 атомов УГЛЕРОДЗ ИЛИ замешанная алкильная группа формулы-(СН 2 Р-у-(сн)(Н 4)-В 5, где Н 4-циклоалкил с 3-6 атомами углерода, К 5-СН 3, циклоалкнл с тремя-шестью атомами углерода. ч у 5 или Ы-0, где Ц - водород, р 1 или 2,чО или 1 О 7.0 д.81.при Ед - водород или низ К. низшая алкильная группа с Штй алкил С 14 атомам углерода,1-4 атомами углерода содер- К водород, ниэшй алкил (за исжащая амно-группу ключением сна), бензилоксикарбонил, Ей - водород или алкильная группа О ацетил или бен 3 нлоксикарбо с 1-4 атомами углерода, НИЛ. Щполучения замещенных аминодикислот общей формулыциклоалил с 3-6 атомам углерода 7 Е - водород, низший алкил с 1-4 атомам углерода, циклоалкил с З-6 атомам угле РОДа или алкоксикарбонил у 5 или 7 Ы-О, где ОвоДор 0 д, ацетил или бензилоксикарбонил, Р 1 или 2, ц 0 или 1,их рацематоа или оптических изомеров или их фармацевтически приемлемъщ солей, обладающих ингибирующей активностью.Известна реакция присоединения азотсодержащих соединений н карбоннльны соединениям. Условия проведения реакции зависят от строения исходных соединений П.Цель изобретеня - синтез новых соединений, обладающих ценными фармакологическими свойствам.Поставленная цель достигается способом получения соединений фор мулы 1, заключающимся в том, что соединение общей формул 11где А, п ц имеют указание значения,К 5 низши алкильньй радикал или амииоалкильньйраднкал аминофункция которого защи 10 ще на такими радикалами как бенвилонснкарбоннл или треттбутилоксикарбонил,К - гидроксилъньй или низший ралкокснльный раднкал 15подвергают реакцинвосстановительно го алкилированияс соединением обтгде На И Кимеют указанные значениядяз с вьщелеиием целевого продукта или со снятием в случае необходимости Ыэащитнык И эфирных групп гидроге- нолизомили омътением. 1ванной соляной кислоты. Нагревают в течение 30 мин на ки 40-пищей водяной бане. Таким образом получают прозрачный раствор, оставляют для установления в реакционной смеси-комнатной температуры н тогда добав ляют 15 мл формальдегида и 3 О мл кон 45 центрироваиной соляной кислоты. Затем нагревают 3 ч при темшературекнпения с обратным холодильником растворителя. Оставлякп охлаждаться, затем осадок отфилътровьшают. После отсасыва- 50 ния его.обрабатывают 200 мл кипящей воды н 30 мл горячего этанола. Раство ры объединяют, затем нейтрализуют добавлением 102-ного аммиака. Тетрагндрохинолин 3 карбоновая 55 кислота кристаллизуется. Кристаллическую смесь оставлякп в течение нот чи в холодильнике, затем осадок отделяют, отсасывают н промваютдэтаиоч лом. Таким обрааом получают 17,3 г неочищенного продукта. Продукт сушат над фосфорным ангидридом под вакуумом. - С,Н.М 02177 Рассчитано, 2 С 67,78 Н 6,26 Ы 7,90. Найдено, 2 С 66,87 Н 6,20 М 7,96. - . ИКспектр 5 МН полоса при 2800.24 ОО см СООТ полоса карбонила приВращательная способность из 1 о 8 (с 221 н ыаон).В трехгорлую колбу последовательно загрйжают 5 г тетрагидроиэохинолин-3 карбоновой кислоты и 30 мл метанола. В эту суспензию добавляют путем приливання избегая превышения температуры О-5 С, 6 г хлористого тионила. Добавление длится около 10 мн. По-окончании добавления под держсиваюг ЩТВРЕМЕШИВЗНИЕ В ТЕЧЕНИЕ. 2 Чкипятят при температуре кипения с обратны холодильником растворителя 2 течение 1,5 ч. После полного раство рения смеси нагрев прекращают, затем выпаривают досуха. Остаток обрабатыт. ВЗЕОТ МЕТЗНОЛОМ ПУТМ ЭКСТРНКЦИИ гриэ Раза И затем доводят досуха. Получают 8 г бесцветных кристаллов, которые очищают путем порошкования с эфиром. Кристаллы отфильтровывают отсасывают их, промывают эфиром н высушивают.Таким образом получают 6,11 г хлоргиц-рата метилчтетрагидронзохинолин 3ткарбокснпата. С 0 НдНО 2 НС 122769. Рассчитано, 2 58,03 Н 6,20 4 Ы 6,15 С 1 15,57. Найдено, Х С 57,79 Н 646 Ы 6,38 С 1 15,67. ИКспектр полоса карбонила при1734 см полоса МН при 28002400 см . Стадия В.6,01 г (О,0264 моль) полученного на предьщущей стадии хлоргидрата растворяют в 50 мл воды и раствор подщелачивают до рН 11 с помощьюМНПН. затем экстрагируют 2 раза Метанол выпаривают ПОД ВЗКУУМОН ВОпо 50 мл серного эфира. Объединенные доструйного насоса н остаток обрабаэфнрные растворы сушат над сульфатом тывают 20 мл воды. После экстракции кальция, фильтруют и выпаривают досу- неоьшшенной части этилацетатом водную ха. Остаточньт сложньт амноэфир 5 фазу подкислиют 4,4 мл 1 нНС 1. Обра(504 г) растворяют в 30 мл диметил- зовавшися осадок экстрагируют 2 раформамнда и этот-раствор добавляют за по 20 мл этилацетата, полученньй к перемешиваемому раствору 5 г раствор сушат над СаО 4, фильтруют н(0,02 б 4 моль) третбутилкарбонил- выпаривают. Полученный остаток пред(5)алаиина в 30 мл диметилформамит 10 ставляет собой искомы продукт,да, охлажденного до О-500. К получен- вес 1,3 г (932). ному раствору добавляют последова 1 СдНН,О 5. тельио 3,6 г (0,0264 моль) 1 окси Рассчитано, 2 С 62,05 Н 6,94,-бензтриазола растворенного в 40 мл Ы 8,04. диметилформамнда, затем 5,45 г 15 Найдено, С 61,64 Н 6,98течение 18 ч при повьщении темпера 20 1,1 г-(0,ОО 316 моль) полученноголбтуры до комнатной. образовавшуюся напредыдущей стадии производного дициклогексилмочевину отфильтровыва перемешивают при 5 С с 4,5 мл безводют и фильтрат выпаривают досуха под ной трифторуксусной кислоты. Полученвакуумом 0,1 мм рт.ст. получая ос ный раствор концентрируют-досуха под таток, которьй снова растворяют в 25 вакуумом 0,3 мм рт.ст. Кристалличес 50 мл этилацетата И снова фильтруют кий гигроскопический остаток последля отделения второй порции днцикло выпаривания представляет собой ценен гексилмочевины. Фильтрат промывают вой продукт в виде трифторацетата,последовательно 80 мл водного насы сольватированного 0,5 моль трифтор щенного раствора ЫаС 1 2 х 40 мл вод- 30 уксусной кислоты, вес. 1,3 г (98). иого 10 тного растворалимонной кис сн 3,г,м 4 о . лоты, снова 80 мл водного насыщенно- Рассчитано, 2 С 45,83 Н 4,21 го раствора ЫаС 1, 2 х 40 мл водного -Ы 6,68. насьщенного раствора ЫаНС 09, водньшк Найдено, 2 С 45,99 Н 4,62 насьщеиным раствором ЫаС 1 до нейт 35 Ы 6,55. рапьной реакции. 0,7 г (0,ОО 19 моль) предыдущего(942) соответствующей свободной аммнокислоты путем пропускания черезОрганичсскую фазу сушат над Са 5 О 4 отфильтровывают и выпариваютъзосузца под вакуумом. Остаток после выпаривании представляет собой целе вой продукт. Вес. 9,1 г (961)тпл 98100 оС (по КОфдерУ) раствора амака. Т.пл. 170 С с разСЮ Нгеыго в ЛОЖЕЕННЕМ. Рассчитано, 2 С 62,97 Н 7,23 Ъ Стадия Е 2(Ы)1 КаР 5 кл 3 ТИЛ Ы 7 ч 73 45 (5)аланил 53 карбокси 1,2,З,4 тетНайдено, 2 с 63,15 н 7,05 дгагидгонтшнфТин- Ы 5797- . 0,849 г (О,0034 моль) 2(5)алач Стадия Г 3 нил 3 карбокснт 123,4 тетрагидроТРЕТ-бУТ 0 КСНКаР 50 ННЛ 2(5)аЛд иаохннолина растворлнт в присутствии ННЛ 3(5)К 3 Рб 0 КСИ 123 э 4 ТТРа ГИд 5 1,9 г (О 0236 моль) пирувиновой кисР 0 Н 3 ОХННОЛИН- 1 лоты при 25 С в 22 мл 1 н.раствора 1,45 Г (000 д НОЛЬ) ПОЛУЧЕННПГП едкого натра и 50 мл буферного растНа ПРЕДЫДУЩЕЙ Стадии Соединения раст- вора с рН 7, взятых израствора, приворяют в 20 мл метанола и к получен готдвленного из 50 Л 0,1 М раствдрад ОМУ РЗСТВОРУ добавляю 44 МЛ 55 первичного фосфата натрия и 29,1 мл (000 д НОЛЬ) 1 Н-ВОДНОГО РЗСТВОРЗ Ед 0,1 н. раствора едкого натра, 0,45 г кого натра Раствор оставляют в те ы(00072 мдль)цианОб 0 РГидрида натрияизбыток цианоборгндрида натрия раэлагаюб добавлением 6 мл К 0 НЦ 9 НТ рированной соляной кислоты. Полученный раствор пропускают через ионообменную смолу (Помех 50 Н). После элюирования дистиллированной водой смолы вплоть до отсутствия НОНЗ хлора продукт, зафиксированньй на смоле вьшывают элюированием с по МОЩЬЮ 1 Л 1 НВОДОГО раствораамММа ка. Аммиачнй раствор концентрируют под вакуумом водоструйного насоса досуха. Остаток после выпаривания представляет собой моноаммачную соль искомого продукта. ПопУЧеННЫЙ вес 0,8 Г (б 9,71)дПолучают по примеру 1 (стадия В) из 3 трет.-бутоксикарбониламино 2Полученный 2 З-трет.бутоксикар боииламнно-2(К 8)метилпропаноил-3 Ч(5)метоксикарбонил 12,34 тетрагидЗ 5 ронэохинолин омыляют с помощью водного раствора едкого натра используя метод примера 1 (стадия Г).(5)карбокси-1,2,3 д-тетрагидроизо хинолин обрабатывают трифторуксусной кислотой согласно методу примера 1(5)карбокси 12,3,4 тетрагидроизо хинолина, которьй превращают в нлор гидрат растворением в избытке 1 Н.0,009 моль НС 1 и 0,515 г 94 ной Пнрувиновой кислоты (ООО 55 моль). Раствор гидрируют под давлением 0,5 бар В присутствии 1 г 102 ногопалладнятна-угле. Около половины теореТНЧеСКОГО КОПНЧЕСТЕЗ водорода ад сорбируются за 1 ч, Суспензиюотфиль тровывают, к фильтрату добавляют 0,515 г пирувиновой кислоты,-затем НЕЙТРЗПИЗУЮТ до рН 77,2 с помощьютриэтиламина. После добавления т г 102 ного палладиянаугле суспензию снова гидрируют под давлением0,5 бар вплоть до исчезновения исходНОГ 0 первичногоамина, ЧТО КОНТРОЛН руют с помощью тонкослойной хроматографии, обнаруживая этот аммн с помощью нингидрида.Реакционную смесь фильтруют И концентрированный фильтрат растворяют в 25 мл воды и пропускают через 125 мл нонообенной смолы (Почек 5 ОН). Фик- сированный на смоле продукт элюнруют 500 мл водного 1 н.раствора аммака. затем 260 МЛ дистиллированной воды. Объединенные элюаты выпаривают досуха. Остаток после вьтаривання представляет собой целевой продукт в ви де моноаммачной соли. Полученньй вес 0,6 г.31,5 г этого индолина (862) получакп омылением в 250 мл 1 н. раствора едкого натра и 150 мл этанола в течеНН 9 18 Ч ПРИ комнатной температуре 43 г (0,224 моль) соответствующего этилового эфира. Воднотспиртовой раствор концентриРУЮТ наполовину, нейтрализуют 25 мл 10 н.сопяной кислоты, образовавшийся ОСДОК ПТфильтРовывают промывают водои и высушивают.Неочищенную кислоту очищают пропусканием через колонку с ионообменной смолой (Почек 5 ОЫ 8 Н) и элюированиМ 2 Н. водным амиаком. Полученную
МПК / Метки
МПК: C07D 209/42, C07D 217/26, A61K 31/47
Метки: аминодикислот, приемлемых, рацематов, или, солей, фармацевтических, изомеров, получения, способ, замещенных, оптических
Код ссылки
<a href="https://by.patents.su/14-479-sposob-polucheniya-zameshhennyh-aminodikislot-ih-racematov-ili-opticheskih-izomerov-ili-ih-farmacevticheskih-priemlemyh-solejj.html" rel="bookmark" title="База патентов Беларуси">Способ получения замещенных аминодикислот, их рацематов или оптических изомеров, или их фармацевтических приемлемых солей</a>
Способ получения производных бензимидазола в виде смеси изомеров или индивидуальных изомеров в свободном виде или в виде их физиологически приемлемых солей

Номер патента: 230
Опубликовано: 30.12.1994
Авторы: Аннамария Уберти, Розамария Микелетти, Массимо Никола, Эрнесто Монтанья, Марко Туркони, Антонио Жакетти, Артуро Донетти
МПК: C07D 487/18, A61K 31/415
Метки: изомеров, смеси, или, бензимидазола, получения, свободном, виде, солей, производных, физиологически, приемлемых, индивидуальных, способ
Текст:
...1 . Эндо 8 метил 8 аэабицикпоЭ 21 177118 С. мс 332 м/е мч 111 окт-З-иловьй эфир в-фтор-дз-дигндраРассчитано, 2 С 61,62 Н 6,39 -2-оксо 1 Нбензимидаэолт 1 карбоновой Ы 12,68. 20 кислоты (соединение 25) Тол 261, СН 2,ЪТ 3 О 4 (ГИДРОХЛОРИТОс . 1 Найдено, С 61 д 5 Н 6,351 Рассчитано,-2 С 54,00 Н 5,38 1112,72, . с - ы 11,81 . Эндо 8 метил-8 азабицикпо 52 С,5 Н 5 Ы 303 НС 1. окт 3 иловый эфир 5 метил 2,3 дигид 25 -Найдено, 2 С 53,65 Н 5,45 ро...
Способ получения фенилалкиламинов или их фармакологически приемлемых солей

Номер патента: 347
Опубликовано: 30.12.1994
Авторы: Карой Можолитш, Йожеф Кнолл, Янош Бергманн, Ева Шомфаи, Золтан Тёрёк, Антал Шимаи, Ева Синньеи
МПК: C07C 211/27, A61K 31/17
Метки: солей, фармакологически, фенилалкиламинов, приемлемых, получения, или, способ
Текст:
...1000 капсул). 30.0 г активнодействующего вещества гомогенизируют совместно с 67.0 г кукурузного крахмала. 50 г продукта Ау 1 се 1 и 50 глактозы. Гомогенную порошкообразную смесь гранулируют с использованием спиртового раствора поливи нилпирролидона на сите Мг 18. сушат и повторно гранулируют на сите Мн 24, После повторного гранулирования добавляют тальк И ПРИГОТОВЛВННЫМИ ТЗКИМ 060330191 гранулами заполняют капсулы Зоар Н Мг 1 либо вручную....
Способ получения производных 1H-1,2,4-триазола или их фармакологически приемлемых кислотно-аддитивных солей

Номер патента: 333
Опубликовано: 30.12.1994
Авторы: Лео Баккс, Ян Херес, Жозеф Мостман
МПК: A01N 43/64, C07D 249/08
Метки: получения, солей, производных, приемлемых, или, кислотно-аддитивных, фармакологически, способ, 1h-1,2,4-триазола
Текст:
...охлаждают до комнатной температуры и продукт экстрагируют 765 вес.ч. бензола. Экстракт промывают 10-ным,раствором едкого натра, высушивают, фильтруют н отгоняют растворитель. Остаток дваждыП р и м е р 2. Согласно методике, описанной в примере 1 применяя эквивалентное количество соответствующего замещенного фенола вместо 4 клор-3-метнлфенола получают следующие промежуточные продукты 1 С 3-бромпропоксиЪ 4-хлор-2 метилбензол, т.кип. 115-11...
Способ получения пиранохинолинов или их фармацевтически приемлемых солей
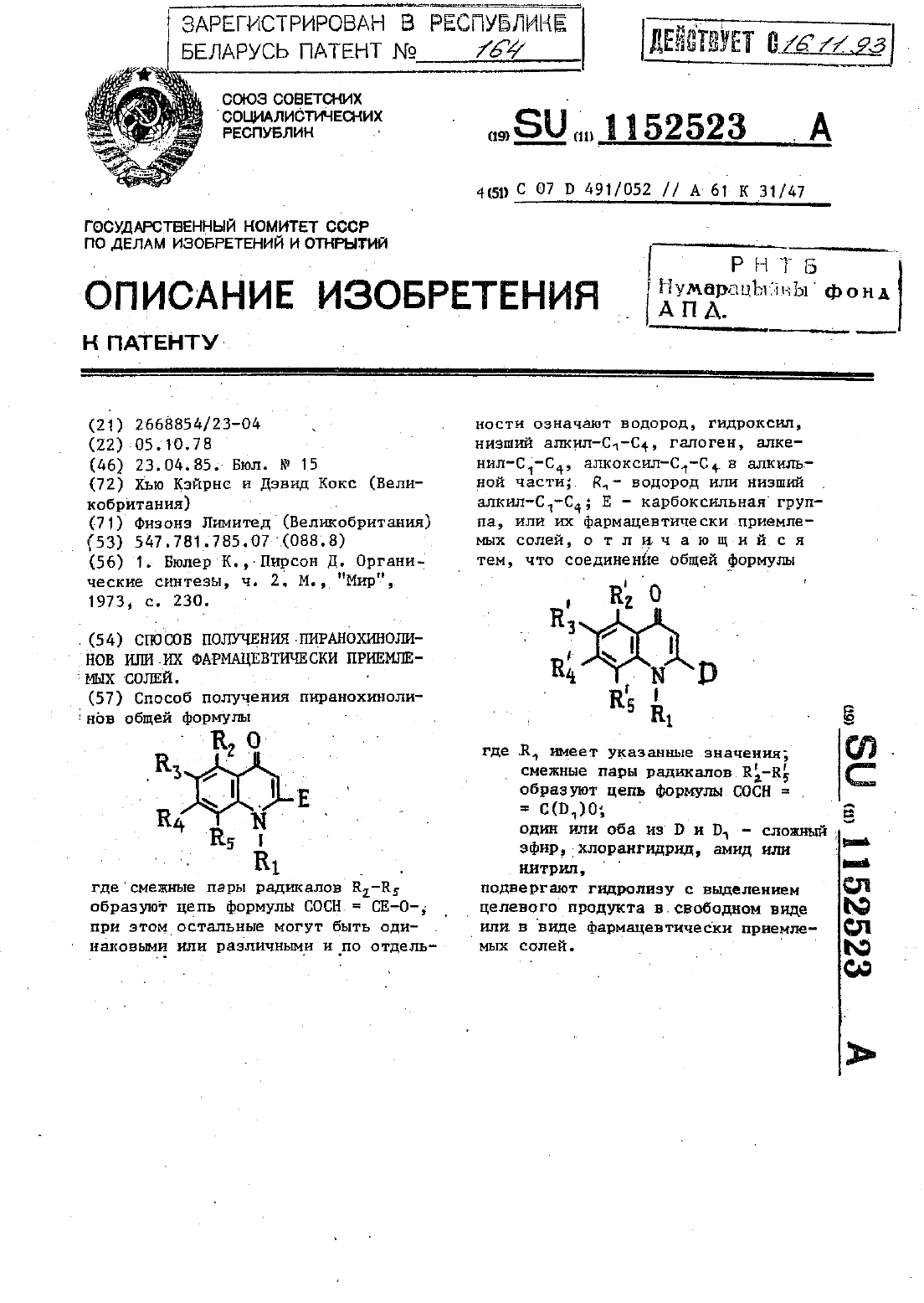
Номер патента: 164
Опубликовано: 30.09.1994
Авторы: Хью Кэйрнс, Дэвид Кокс
МПК: A61K 31/47, C07D 491/052
Метки: получения, фармацевтически, приемлемых, или, солей, способ, пиранохинолинов
Текст:
...л.). СТРУктУра проДкта подтверждена масс и ЯМРспектром. Ъ. Динатрий 4,6 диоксо 10 пропил 4 Н 5 НПИРаН 0(332-)хинолин 28 ч дикарбоксилат. бис-Киспоту стадии 1(135 О абикарбонат натрия (О,661 гв воде(150 мл) иагреваютд перемешиают до тех пор, пока раствор станет прозрач ны.Этотрастворфильтруют,афильтрат(90,д г) перемешивают в сухом диметилформамиде (500 мл) в течение 17 Ч Реакционную смесь слиают В воду и продукт экстрагируют эфиром....
Способ получения хинолинкарбоновых кислот или их фармацевтически приемлемых солей

Номер патента: 352
Опубликовано: 30.12.1994
Авторы: Петер Ритли, Мария Балог, Иштван Хермец, Агнеш Хорват, Делле Вашвари, Геза Керестури
МПК: A61K 31/47, C07D 401/04
Метки: фармацевтически, или, солей, получения, способ, приемлемых, хинолинкарбоновых, кислот
Текст:
...Найдено, С 61,58 Н 5.50 М 12.61.(ацетато-Ш-бора и М-метилпиперазина согласно примеру 1 получают 1-циклопропипб-фтор-7-(4-метилпиперазин)1 ,4-ди гидрод-оксохинолин-З-карбоновую кислоту. Про дуктреакции разлагается при 248-25 ОС.П р им е р 3. В 16 мл диметилсульфоксида нагревают до 90 С при перемешивании 4.1 г (1-циклопропил-6-фтор-7-хлор-1 А-дигидро-д-оксохинолин-3-ка 5 боксилат-03.О )-б.ис(ацетато-9-бора и 3,7 г М-зтилпиперазина. После 10...
Предыдущий патент: Способ производства цементного клинкера
Следующий патент: Способ получения 4-ацил-2,3-дигидро-1,4-бензоксазинов или -бензтиазинов
Случайный патент: Антифрикционная полимерная композиция