Способ получения 2-окси-4-(метил-тио)-масляной кислоты
Номер патента: 376
Опубликовано: 30.03.1995
Авторы: Деннис Артур Руст, Масахару Такано, Лоренс Рассель Вольф









Текст
Изобретение относится к усовер шенствованному способу получения 2 окси 4-(метилтио)-масляной кисло ты (НМВА), которая может быть использована как добавка к корму, в частности, для домашней птицы.ЦЕПЬЮ НЗОбРЕТгНИЯ ЯВЛЯЕТСЯ ПОЗЫ ШеННЕ КЗЧЕТНЗ ЦЕЛЕВОГО ПРОДУКТЗ, 10 меньшую вязкость н лучшую теплостой.кость что достипается гидролиэом 2-окси-4(метилтио)-бутиронитрила(НИВЫ) сначаца 50 т 1 О-ной серной кислотой при 25-65 С с последующей обработкой полученного при этом 2 окси-д-(метилтио)-бутирамица 30 502-ной серной кислотой приВ 9-120 д экстракцией с несмешивающимся с водой органическим растворителем и реэкстракцией целевого продукта в присутствии воды в количестве 5 12,2 мао.2 в пересчете на экстракт.П р и м е р 1. НМВЫ (13210 Г,95 ной чистоты после газовой хроматографии), полученный из метилмеркаптана акролеина и цианистого водорода, добавляют к 50 ному по весу водному раствору серной кислоты (1964 г) при 5 ОС в течение 30 мн 3 в колбу с рубашкой, снабженной мешалкой, емкостью 1000 мл. Полученную смесь выдерживают еще 30 мн при 50.0. Промежуточный гидролизат быстро нагревают до 90 С Св течение 20 мин) И подвергают опять реакции В течение 100 мин при 90 С. Через 13 мин при 90 С происходит разделение фаз, где органический слой, содержащий НМВА, выделяется в.виде соли. По завершении реакции гидролиза, добавляют 28-ный по весу аммиачный раствор (5897 г) к гидролизату при 80 С в течение 20 мин. После добавки немного более чем половины аммиачиого раствора из водной фазы осаждаются мелкие кристаллы. В конце добавки аммиака при значении рН 1,76 из-за сильной кристаллизации бьшо очень трудно перемешивать смесь.Для выделения НМВА из побочных продуктов, содержащихся В нейтрализоВЭНОМ ГНДРОЛНЭЗТЭ, ПРИМЕНЯЮТ триВ первом из этих способов нейтрализованный гидропизат (50 млб 3 Г) смешивают с метилпропилкетоном(50 мл) и водой (10 мл) для экстрагирования НМВА из водной в органиче 55скую кетоновую фазу. Кристаллы сульфата аммония-остаются в водном слое. Обе фазы подвергаются анализу, ре эультаты которого приведены в табл.1...п--.-ь..-ц- Растворитель упаривают из органической фазы под вакуумом при 70 С в течение 60 мн, затем давление па цра снижается до 16 мм рт.ст. абс.88-ный по весу раствор НМВА в водеимеет цвет, которьй на шкале Гард- нера показывает значение 5.Согласно второму способу получения нейтрализованный гидролизат(50 мл) смешвают с метилпропилкетоном (50 мл) с цепью экстракции НМВА. После смешения гидролизата и растворителя разделение фаз становится трудным изчза высокого содержания твердых веществ. По завершении разделения после отстаивания в теЧВНИЕ НОЧИ ОРГЗННЧВСКУЮ И ВОДНУЮ фа зы подвергают анализу, результаты которого приведены в табл.2.3 1428193 ц Растворитель-упаривают из органического слоя под вакуумом при 70 с в течение 60 мин, причем понижается давпеннеЪара до 16 мм рт.ст. про 5 дукт НИВА на дне анализируют, он содержит-74,9 вес.2 мономера НИВА,-воды. Значение цвета 88-иого раствора продукта НМВА в воде на шкале Гарднера составляет от ддо 5. ц В третьем способе разделении ней снабженный пропеллерной мешалкой. Полученный раствор вьщерживают еще в течение 30 мн, затем температуру Реакции повышают до 90 С н течение 26-30 мин и вьщерживают при 90 Св течение 120 мн. По окончании реакции порцию гидролизата (16044 г смешивают с метилпропилкетоном(12835 г) при 50-60 С в разделитепьном сосуде емкостью 5 л В тече нне Окд 10 мин, чтобы экстрагиротралнзованиы гидропиэат дистилли- вать продукт НМВА из гидролизата.руют от летучикпод вакуумом при 7 ОЧ 3 -затем водную фазу удаляют из сосуда в течение 60 мн, причем давление па-15 Н слой экстракта(2 О 73,2 г) промыеч. ра снижается до 15 мм рт.ст. абс вэютЬодой (2 О 75 г) при 50 с,-вод В перегонном сосуде образуется очень ный сддй(д 8,8 Г 5 о 2 НМВА) Былинагустой шлам. Отфнльтровав твердые ют из сосуда3 ЩеСТ 33 гФидЬТаТ анадИ 3 гУЮТ- он Раствор упаривают из экстракта содерлит 75,2 вес.2 мономера нива, 20 под вакуумом при 50 оС продолжая 2 О,2 вес.олигомеровНМВА 14328 вес.2 перегонку ока давденепара не воды. Значение цвета 88-ного по ве- снизилось до 30 мм рт.ст. Затем н су раствора прводукта НМВА в воде остатку под поверхностью добавляют на шкале Гарднера составляет от 4 вдду (20 мл) И температуру В дере д 9 5- гонном сосуде повьшают до 70 С что П Р И Мое Р 2 НИВЫ (2003 ПОЛУ бы отгонять остаточный растворитель чениыи по способу примера 1 медпен- с водяным паром. Когда давление но добавляют к БОЙ-кому по весу раст-Ч пара снижается до гоммртстабс ФРГ вору серной КИСЛОТЫ (299 Г) при 50 С 70 С дистилляция с водяным паром В ТЕЧЕНИЕ 30 МИН В КПбУ С РУб 3 ШКЙ 30 закончена. Чистый продукт, оставемкостью 1000 мл. Полученную смесь шмйсн в перегонном сосуде, после БыдеРЖи 3 аЮТ еще В Течение 30 ММН- ПО дистилляции с водяным паром подверлученни промежуточным гидролизат- гают анализу. он содержит 74,0 Вес.быстро нагревают до Эоас (В течение могомера НМВА 24 4 вес 7 опигоме о 20 мн) и выдерживают еще в течение 35 НМЗА 1 8 вес 7 Езды И Ь 45 вес 7 р В 1 3 от .вРолизат прнобгчтагт коричневатый бавляют во ой ЬЬ. ПКЙРЗцвет. Конечньт гндролизат состоит из д лучая нь .п весу продукт НИВА, цвет которого надвух фаэ. . шкал Г Не подвергая гидролизатнейтра- 49 до бе арднера имеет значения т 5ливации, его смешивают с одинаковым количеством метилпропилкетонаи пос ле разделения фаз растворитель ОТ гоняют в вакууме от экстракта прн 703 в течение 120 мин. Полученный про.дукт содержит 63,6 вес..мономера НИВА, 35,2 вес. олигомеров НМВАП ри м е р 4. НИВЫ (26316 г), получеиныйспособом примера 1, мед 45 ленно добавляют к 652 ному по весу раствору серной кислоты (30145 г) при 50 С в течение 60 мин в колбуполученный по способу примера 1, при0,11 вес. НМВЫ, 0,61 вес.2 промежуточного амида, 211 вес.2 воды и 0,27 вес.2 сульфатных ионов. Цветт 882-ного водного раствора продукта на шкале Гарднера имеет эначение от 5 до 6.к 502 ному водному раствору серной кислоты (981 г) при 50 С в течениес рубашкой емкостью 1000 мл, снабженную мешалкой. Полученную смесь выдерживают в течение 30 мин при 50 С. Затем к промежуточному гидролизату добавляют воду (18891 г),чтобы снизить концентрацию гидролизующей кислоты до 40 по массе на основе отсутствия нитрила. Температуру содержимого реактора затем повышают с 50 до 90 С (в течениеворителя определяют коэффициенты расНа первой стадии гидролиза т.е. во время реакции) в 65 ном растворе серной ниспоты (начальная концентрацня при 50 С) вязкость реак 5 ционной смеси сипъио повышается и реакционная система начинает образовать две отдельные фазы, одна из них содержит промежуточный 2 окси-4-метилтиобутирамид а другая т только что добавленный к смеси НМВЫ. Во время-второй фазы гидролиза,т.е. в ходе превращения промежуточного амида в кислотный продукт при температуре 90 С, сохраняется одна фаза без всякого разделения фаз. По окончании гидролиза гидролизат подвергают анализу, он содержит 35,21 вес. мономера НМВА,0,31 вес.2 димера НМВА, 0,01 вес.НМВЫ и 0,01 вес. промежуточного амида. Другую порцию гндролизата НИВА данного примера подвергают экстракт ции, применяя различные растворители. В каждом примере 100 вес.ч. гидролизата смешивают с 60 вес.ч. растворителя в разделительном сосуде. После перемешивания и разделения фаз 100 вес.ч, органического слоя промывашт 12,5 вес.ч.воды и водный рафинат (100 вес.ч.) промывают 60 вес.ч. растворителя. Всеэкстракции проводят при комнатной температуре,т.е.при 25 С.Для каждогораст 10пределення при равновесии между орт ганической и водной фазами. Коэффициент распределения определяют по р концентрации НМВА в органической фа.С РаЗЙНЧНЬЕМИ РЗСТВОРНТЕЛЯМН В ДЗННОМ примере приведены в табл.З. А л Т а бцл и ц а 3Пр и м е р 5. Получают НМВА по следующей схеме. В этой системе гид ролизат НМВА получают в периодической системе реакции, состоящей из одиночного реактора, снабженного мешалкой,однако на двух стадиях реакции. На первой стадии НВЫ медленно добавляют к серной кислоте, причем получают промежуточный гидролнэат содержащйи температуру повышают, чтобы превратитъ промежуточньй амид в НИВА. 0 кон- 5 чательньй гндролизат подают в промежуточный сосуд. Оттуда его непрерывно подают приблизительно в середину экстракционной колонны Карра с сит в которую растворитель подают воэле дна, а проиывную воду возле верха. Верхний погон экстракта предварительно нагревают в теплообменнике Н подают В колонну для дистилляции с водяньм паром. На ни .5 зу колонны находится жидкий продукт,содержащий НМВА н воду. Пары из верхней-части колонны конденсируются в конденсаторе и направляются в сепаиз которого растворитель рециркулируется в нижнюю часть экстракционной колонны, а вода рециркулнруется в верхнюю часть экстракционнойВьжодящшй из низа экстракционной колонны рафинат подвергают дистилляцн с водяным паром в колонне, чтобы выделить остаточный растворитель из паров в верхней части колонны,которые также направляются к конден 25сатору где они конденсируютсяиподаются в сепаратор. На дне колонНЫ НЗХОДЯТСЯ ВОДНЫЕ ОТХЬДЫ, КОТОРЫЕВ типичном периодическом гидролизе данного примера 65,1 мас. серной кислоты (1 д 2,3 кг) подают в реактор на стадии 1 и в реактор медленно добавляют НИВЫ (120,1 кг) в течение 61 мин при температуре от 50 до 5 дС-до На стадии 1 А промежуточный гднроли-Г эат разбавляют до концентрации кисло 35тты 40,1 (на основе отсутствия нит рила), добавляя воду и нагревая до-ролиэат еще выдерживают при 90 С втечение 75 мин. Затем удаляют летут ЧИЕ КОМПОНЕНТЫ, ПОСТЕПЕННО СННЖЗП давление примерно до 110 мм рт.ст.абс. в течение ок. д 5 мин, снижаятемпературу примерно до 65 С. При- 50 блиэительно 11 кг вещества улетучиваются. Затем гидролизат вливают В промежуточньй сосуд.Конечный гндролнзат из промежу- 55точного сосуда непрерывно подают в колонну в количестве 181 г/мин и метилиэобутилкетоновый растворитель СМВК) подают в нижнюю часть зкстрак-, ЦНОННОЙ КОЛОННЫ В КОЛИЧЕСТВЕ100 г/мин. Промывную воду вводят в верхнюю часть колонны. В колонне ПРОВОДНТ НСПРВРЫВНУЮ ЩТВОТИВОТОЧНУЮ ъ экстракцию примерно при 59 С при БОЗВРЗТНОПОСТУПЗТВПЬНОМ ДВИЖЕНИИ тарелок с 140-228 ударам в минуту,получая экстракт, который удаляют из верха колонны, н водный рафинат,который Удаляют из низа колонны.Предварительно нагретый в теплообмен ннке экстракт подают а колонну для перегонки с водяны паром, где дистиплируютрастворитепь при давлекии 235 Н ртдст. в верху, при 82 С в верхней части колонны и 88 С в нижней части колонны, получая низовой продукт в количестве 78 г/мн, который состоит из водного раствора НМВА. Пары на верхней части, содержащне 100 г/мн М 1 ВК н 50 г/мин воды, конденснруются в конденсатор и поступают в сепаратор.Рафннат нэниэа колонны подвергафне при давлении в верхней части колонны 760 мм ртст., температуре вер ха 97 С и температуре в сосуде107 С, получая пар вверхней части,содержащй 0,9 г/мин НЕВК и 5 г/мн воды, которые смешивают с парам из верхней части колонны, конденсируют в конденсаторе и направляют 5 сепаратор. При дистилляции рафнната в колонне в нижней части колонныполучают продукт 144 г/мин, который направляют к отходам. Экстракционнан колонна представляет собой.колонну Керра с ситчатой тарелкой диаметром 2,54 см нвысотой 2,1 м. После достижения стабилизированното состояния гидролиэат, поступающий из сосуда, и водный продукт из нива ПВЬЗЬОННОЙ КОЙОННЛ ПЭРНОДИЧЕСКН- От бнрают для анализа. Результаты анализов приведены в таблд. 1 п
МПК / Метки
МПК: C07C 149/20
Метки: 2-окси-4-(метил-тио)-масляной, способ, получения, кислоты
Код ссылки
<a href="https://by.patents.su/9-376-sposob-polucheniya-2-oksi-4-metil-tio-maslyanojj-kisloty.html" rel="bookmark" title="База патентов Беларуси">Способ получения 2-окси-4-(метил-тио)-масляной кислоты</a>
Способ получения кальциевой соли (-окси-(-метилмеркаптомасляной кислоты

Номер патента: 341
Опубликовано: 30.12.1994
Авторы: Роберт Майкл Виджилент, Стивн Ервин Глайч, Ерл Весли Камминс
МПК: C07C 149/20
Метки: соли, окси-(-метилмеркаптомасляной, способ, кислоты, кальциевой, получения
Текст:
...предпочтительно используют небольшой избыток кальцневогосоединения Предпочтит тельны является избыток-не выше, чем 102, в противном случае снижается чистота продукта.Для получения кальциевой соли раствор кислоты предпочтительно добавляют прямо к сухой окиси или гидроокиси кальция в смесителе для твердых веществ.Если кислоту добавляют прямо к окиси или гидроокиси кальция, полученная реакционная масса становится очень вязкой и ее трудно...
Способ получения N-[2-(4-фторфенил)-1-метил]-этил-N-метил-N-пропиниламина в виде рецемата или L-изомера, или их солей

Номер патента: 343
Опубликовано: 30.12.1994
Авторы: Ева Шомфаи, Золтан Эчери, Йожеф Кнолл, Золтан Тёрёк, Карой Можолич, Ева Синньеи
МПК: A61K 31/135, C07C 211/27
Метки: виде, получения, рецемата, или, n-[2-(4-фторфенил)-1-метил]-этил-n-метил-n-пропиниламина, солей, l-изомера, способ
Текст:
...раствором хлористого водорода. Выпадающий в осадок продукт реакции фильтруют и высушивают. Таким образом получают 2.2 г-М-метил-Е-д-фторфенил)-1-метил- этиламина и 1.46 г(0.00973 моль) Е-винной кислоты растворяют. в 15 мл этанола. Полученную смесь охлаждают до -1 ОС И кристаллируют в течение б - В ч.Кристалли 1.15 г солит.пл. 88 - 94 С. . Полученную соль (05 г) суспендируют в 3,2 мл воды и добавляют 1.3 мл 1 О-ного раствора гидрооксида...
Способ получения N-[2-(4-фторфенил)-1-метил]-этил-N-метил-N-пропиниламина

Номер патента: 351
Опубликовано: 30.12.1994
Авторы: Ева Синньеи, Ева Шомфаи, Золтан Тёрёк, Карой Можолич, Золтан Эчери, Йожеф Кнолл
МПК: C07C 85/08, A61K 31/135, C07C 87/28...
Метки: способ, n-[2-(4-фторфенил)-1-метил]-этил-n-метил-n-пропиниламина, получения
Текст:
...а затем по каплям Й ВыПаРИВаЮТ- 0 СТ 3 Тк растворяют при перемешивании добавляют 60-нът В 101 НЙ дЯй киТе Й экстра раствор 7,56 г (ОО 45 моль) пропарГИРУЮТ 5 Н 3 д- 3 дЫй й дщ гилБромидав толуоле. Реакционную дава И Экстрагируют 5 е 3 л смесь перемешивают в течение 3-4 ч ПОСП ЧЕГО 5 еН 3 дьый экстракт вы 50 при 354 ПС, отфильтровывают, промыСУШИВдЮТ И выпаРнваЮТ ОТаткдереТ вают ацетоном и фильтрат выпариваГПНЯЮТ ПОД ЕаКУУМ 0 М- ПОЛУЧНЮТ...
Способ получения смешанных ангидридов хинолинкарбоновой кислоты и борной кислоты
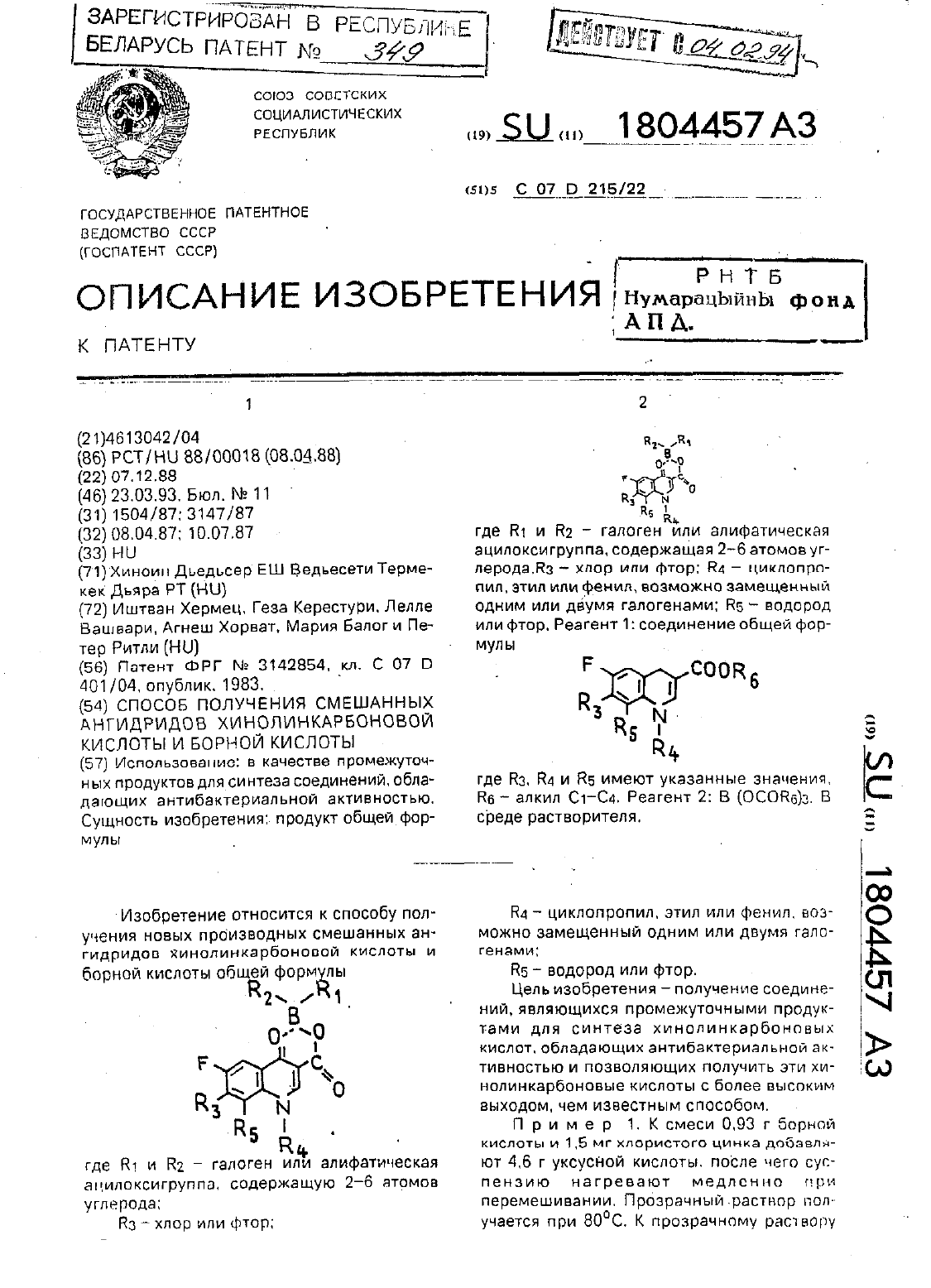
Номер патента: 349
Опубликовано: 30.12.1994
Авторы: Иштван Хермец, Геза Керестури, Агнеш Хорват, Петер Ритли, Мария Балог, Лелле Вашвари
МПК: C07D 215/22
Метки: борной, кислоты, хинолинкарбоновой, ангидридов, смешанных, получения, способ
Текст:
...при охлаждении в течение 3 ч. Выпадающие в осадоккристаллы фильтруются. промываЮТСЯ ВОДОЙ И МЕТЭНОЛОМ И ВЫСУШИВЕЮТСЯ. Получают 7.7 г 1-этил 6.7.8-трифтор-1.4-дигидро-д-оксо-хинопин-З-карбоксипат-дзр бис(ацетато-0)-бора Продукт реакции разлагается при 211 С.Анализ для формулы С 16 Н 13 ВР 3 МО 7П р и м е р. 5. К 30 мл б 0-ного водного раствора тетрафтороборной кислоты при перемешивании добавляют 3.64 г(0.01 моль) этилового эфира 1 -4...
Способ получения , -дифенил-4-арил-4-окси-1-пиперидинбутанамид-N-оксидов или их стереоизомеров

Номер патента: 331
Опубликовано: 30.12.1994
Авторы: Людвиг Пауль Кауманс, Лауренс Валс
МПК: C07D 211/04, A61K 31/445
Метки: способ, или, получения, дифенил-4-арил-4-окси-1-пиперидинбутанамид-n-оксидов, стереоизомеров
Текст:
...1 пиперидинбутанамидад 12 г 5 Оного раствора перекиси водорода,200 г метанола и 320 г метилбензола(ЖХВД) над силикагелем используя в качестве элюента смесъ из трихпорме тала, метанола И метанола, насыщенного аммонием (909 О 1 по объему). Чисе тые фракции собирают и элюент выпарит вают. Остаток кристаллизуют из смеси2,2 оксибиспропана и небольшого ко ЛИЧВСТВЗ МЕТЭНОЛЗ. ПРОДУКТ ОТФИЛЬТРОП р и м елр 6. Вперемешиваемый раствор из 20 г...
Предыдущий патент: Устройство для очистки сельскохозяйственной продукции
Следующий патент: Пакет для ядохимиката
Случайный патент: Устройство для дражирования семян