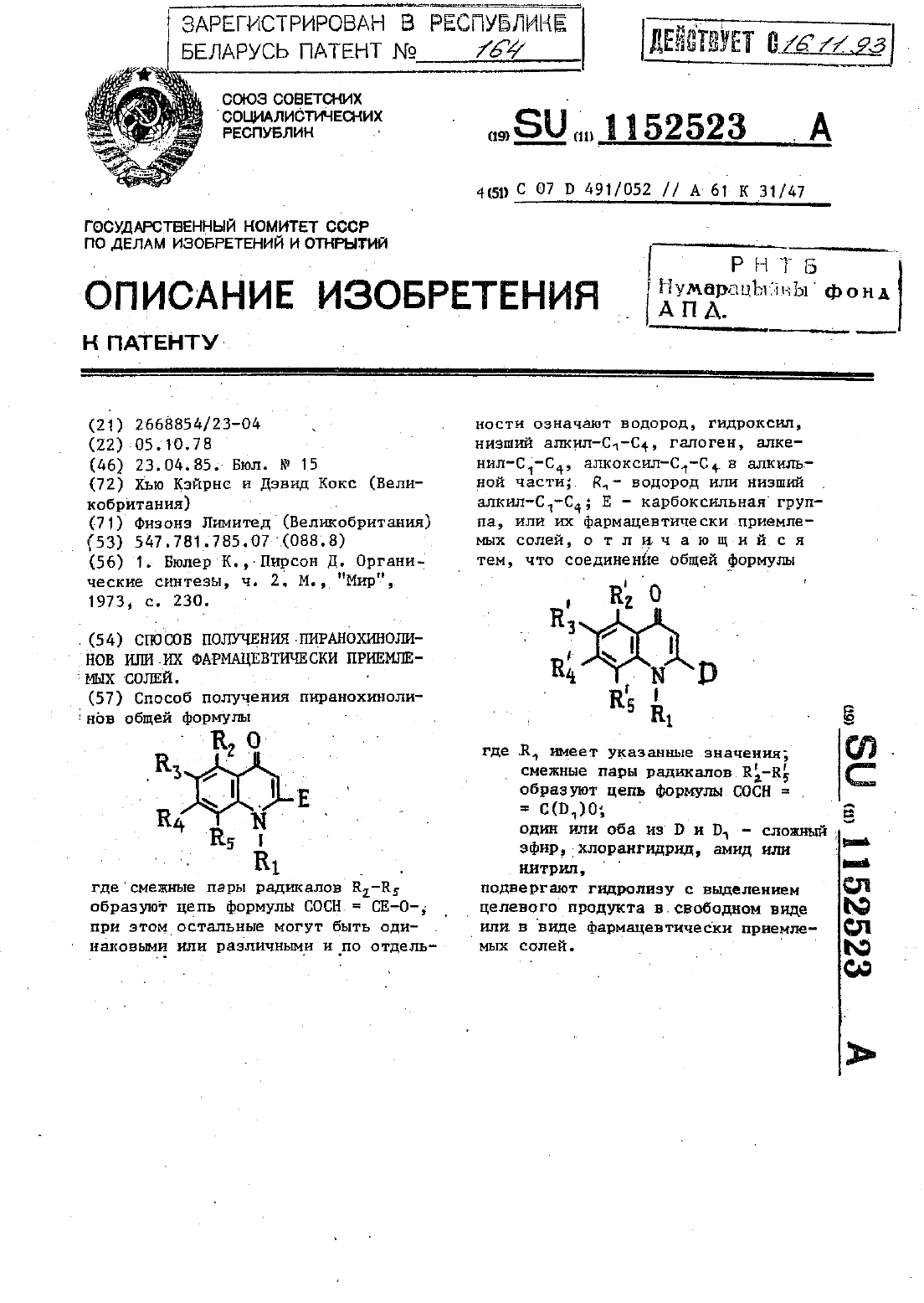
Способ получения пиранохинолинов или их фармацевтически приемлемых солей




Текст
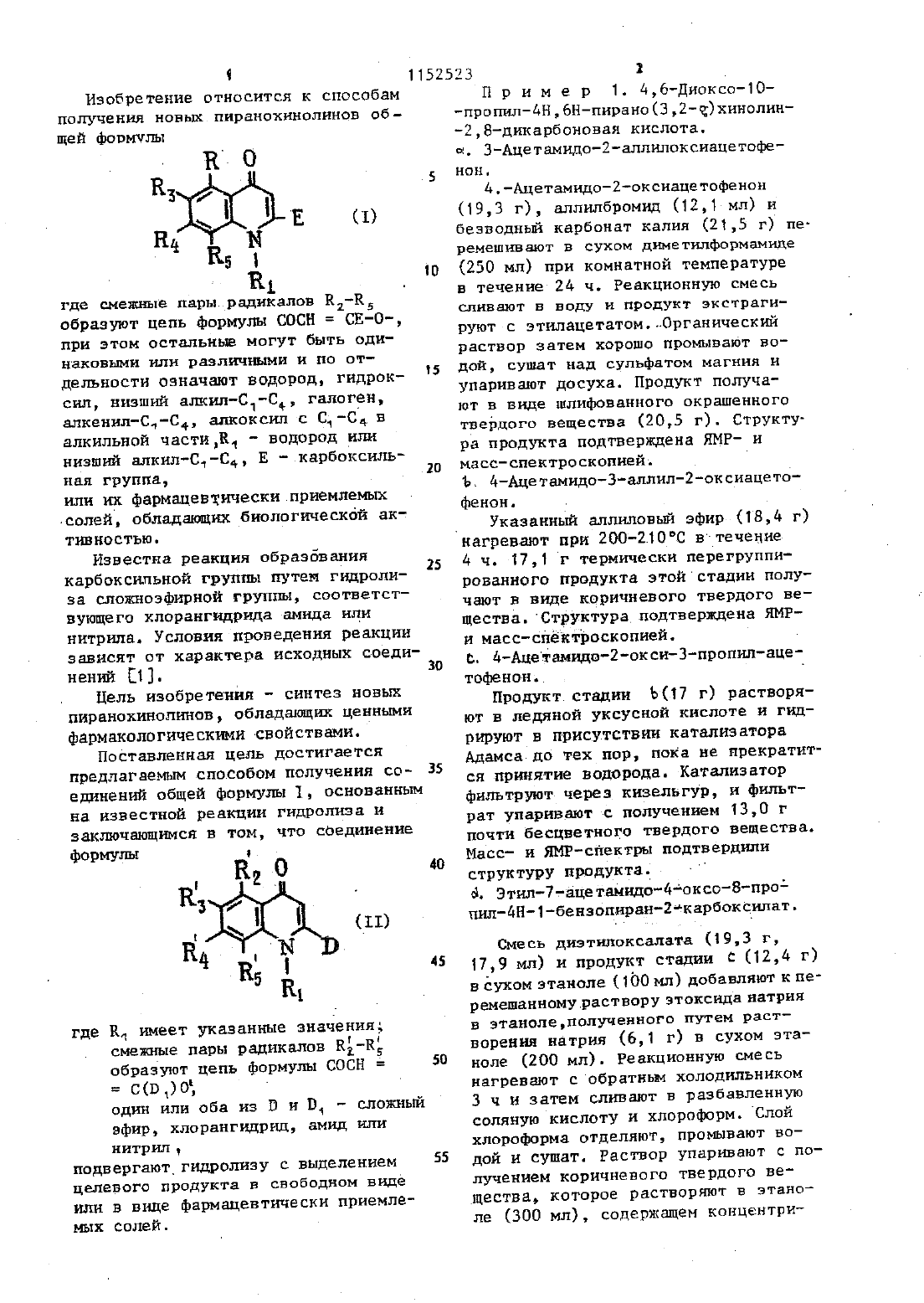
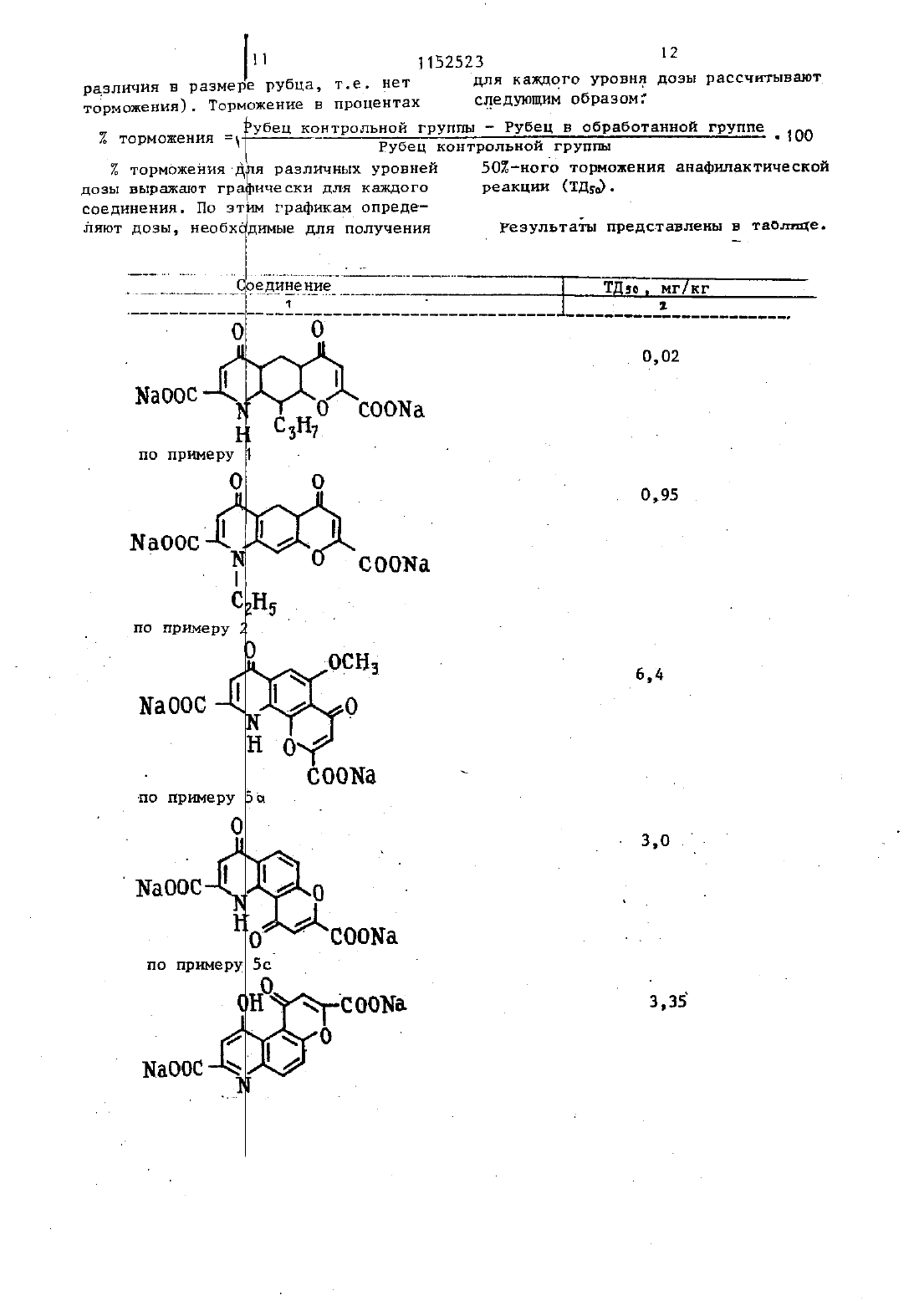
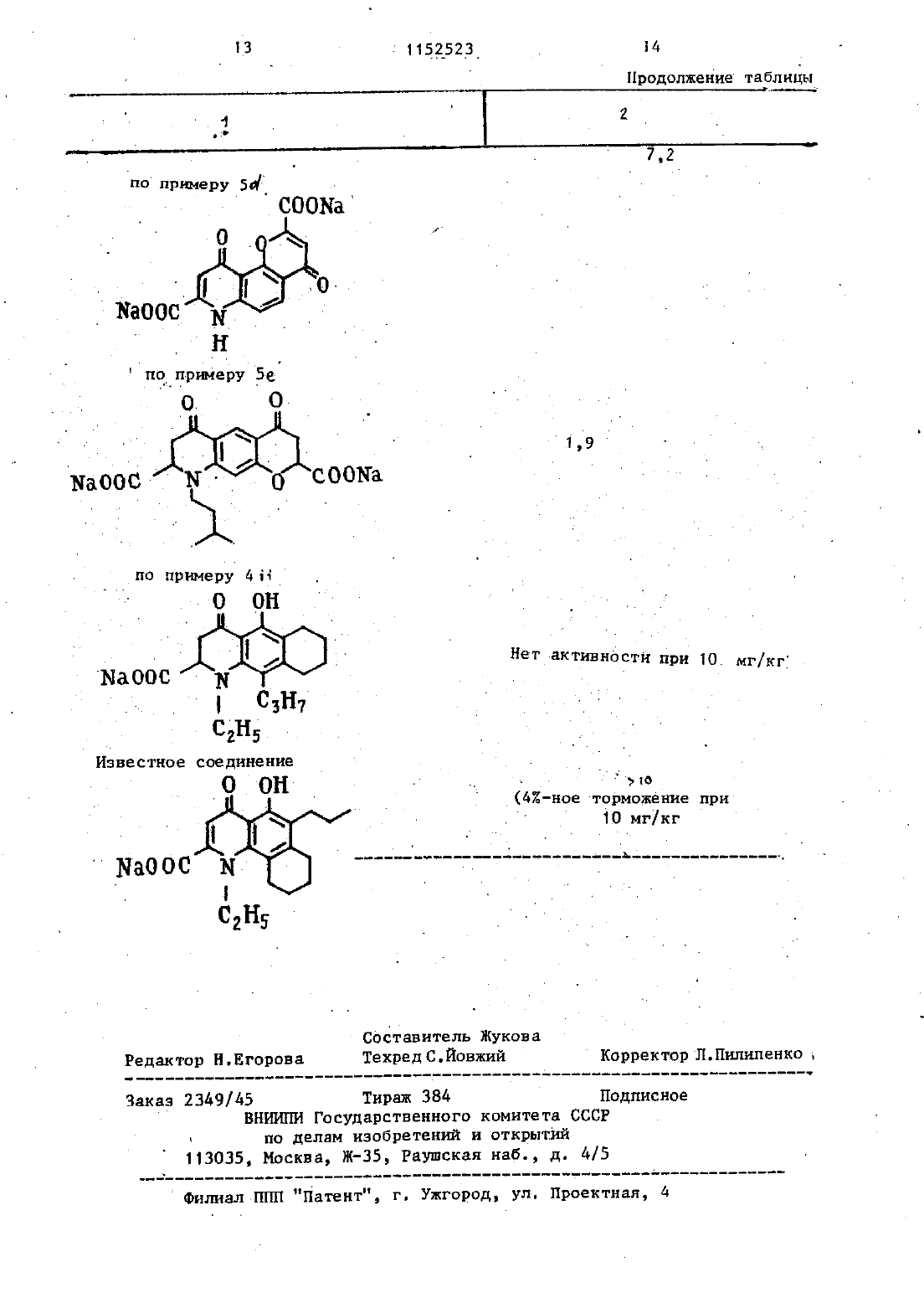
Изобретение относится к способам получения новын.пиранохинолинов об щей формул Т- С)1 . где смежные пары.радикалов Е 1-ЕЕ образуют цепь формулы СОСН СЕ-О-,при этом остальные могут быть одиНЕКОВЫИ ИЛИ ВЗЗЛИЧЫМИ И ПО ОТ делъности означают водород, гидроксил, низши алилС 1-Ср, галоген,алкенилС-С 4 алкоксил с Ст-С 4 в алкильной частиК - водород или низший алкил-С,-С Е карбоксильная группа, . или их фармацевтически.приемпемыхИзвестна реакция образования карбоксильной группы путем гидролиза сложноэфирной группы, соответствующего кпорангидрида амида или нитрила. Условия проведения реакции зависят от характера исходных соединений 1.фармакологическимисвойствами. Поставленная цель достигается предлагаемым способом получения со 35 единени общей формулы 1, основанным на известной реакции гидролиза и заключающимся в том, что соединениеформулы на О 40 гр . 3 (11) ВЧ Ан д 45 11 где Ед имеет указанные значения смежные пары радикалов Ка-Кобразуют цепь формулы СОСНодин или оба из В и В, - сложныйнитрид,подвергают гидролизу с выделением целевого продукта в свободном виде или в вице фармацевтически приемлемых солей. е 50(193 г), аллилбромид (12,1 мл) и безводньй карбонат калия (21,5 г) перемешиают в сухом диметилформамиде(250 мл) при комнатной температуре в течение 2 д ч. Реакционную смесь СЛНВЭЮТ В ВОДУ И ПРОДУКТ ЭКСТРЕГН руют с этилацетатом.Органический раствор затем хорошо промывают водой, сушат над сульфатом магния и уваривают досуха. Продукт получают в виде шлифованного окрашенного твердого вещества (205 г). Структура продукта подтверждена ЯМР- и масс-спектроскопией.Указанный аллиловй эфир (184 г)д ч. 17,1 г термически перегруппированного продукта этой стадии получают в виде коричневого твердого вещества. Структура подтверждена ЯМРи масс-спектроскопней. С- д-Ацетамидо-2-окси-3 пропил-ацетофенон. 0 Продукт стадии Ъ(17 г) растворяют в ледяной уксусной кислоте и гидрируют в присутствии катализатора Адамса до тех пор, пока не прекратится принятие водорода. Катализатор филътруютчерез киаельгуР И фильтрат упаривают с получением 13,0 г почти бесцветного твердого вещества. Масс- и ЯМР-спектры подтвердили структуру продукта 6. Этил-7-ацетамидо-д-оксоВ-пропил-4 Н-1-бензопиран-гтнарбокснлат.Смесь диэтилоксалата (19,3 г,17,9 мл) и продукт стадии С (124 г) в сухом этаноле (100 мл) добавляют к перемешанномурастворузтоксиданатрнд в этанолеполученного путем растворения натрия (6,1 г) в сухом этаноле (200 мл). Реакционную смесь нагревают с обратным холодильником 3 ч и затем сливают в разбавленнУЮ соляную кислоту и хлороформ. Слой хлороформа отделяют, промывают водой и сушат. Раствор упариают с получением коричневого твердого вещества которое растворяют в этаноле (300 мл), содержащем концентри3 рованную соляную кислоту (З мл), И .4 все это нагревают с обратньм холо д дильником 1 ч. Реакционную смесь сливают в воду, а продукт зкстрагит РУТОТ В ЭТИПЗЦТЗТЕ, ЗЗТЕМ ПРОМДЫВЕЮТ водойзтсушат.Растворительупаривают сполучением 1 О 15 липкоготвердоговеш Ществакоторое имеет структуру ожидаемого вещества (данные масс И ЯМР-спектроскопии).Раствор амида стадии ЫСЧО г) в этаноле (300 мл), содержащи концентрированную соляпую кислоту(5 мл), нагревают с обратнм холодильниом 8 ч. Реакционную смесь разбавляют водой и экстрагируют в этилацетата. Экстракт промвают водой, сушат, раствор упаривают с получением темного коричневого полутвердого продукта. Его подвергают хроматографии-на силикагеле с использованием эфира в качестве разбавителя. Получают 4,8 г требуемого продукта. Структура подтверждена масс И ЯМР 4 спектром т.пл. 8 д 87 С. Е. 8-ЭтоксикарбоНил 2 метоксикарбо нил 46 диокси 70 пропил-4 Н,6 Нпира 20(1,2 д г 01 Мл) нагревают В ЭтаН де (30 мл) в течение 26 ч. РеакЦИ 0 Н аую смесь охлаждают ДО ОС Н 9 Р 8 СТ воримое желтоткоричневое твердое вещество собирают ПРИ ПОМОЩИ ФИЛЬТ рации, промывают небольшим количеством этанола И сушат с ЦОЛУЧЭНИЭМ 2,0 г продукта, который является смесъю малеинового И ФУМЗРОБОГО 9 ФИ ров, полученных путем добавления по Мишелю амина К ацетилену.полифосфорнои кислотой (80 мл)и нагревают на паровой бане при перемег шиаини в течение 20 мин. Реакционную смесь сливают затем на лед и перемешивают с этилацетатом. Органичес 5 д кий слой отделяют, промывают водой и сушат. Растворитель упарнвают с получением 1,6 г желтооранжевого Твердого вещества. При перекристаллнэации этого твердого веществаиз этилацетата получают требуемый продукт в Виде пушистых оранжевых иголок, т.пл. 187188 С.(100 мл) Н воде (50 мл в течение 1,5 ч. Все это сливают в воду И подкисляют с осаждением ъжелатинового твердого вещества. Его собирают при помощи фильтрации, нагревают с обратньм холодильником с этанолом, И продукт отделяют центрифугировааием(Е,4 г тпл.3 о 3-3 о 4 с (ра 3 л.). СТРУктУра проДкта подтверждена масс и ЯМРспектром. Ъ. Динатрий 4,6 диоксо 10 пропил 4 Н 5 НПИРаН 0(332-)хинолин 28 ч дикарбоксилат. бис-Киспоту стадии 1(135 О абикарбонат натрия (О,661 гв воде(150 мл) иагреваютд перемешиают до тех пор, пока раствор станет прозрач ны.Этотрастворфильтруют,афильтрат(90,д г) перемешивают в сухом диметилформамиде (500 мл) в течение 17 Ч Реакционную смесь слиают В воду и продукт экстрагируют эфиром. Органически раствор затем промывают хорошо водой, сушат над сульфатом магния и упаривают досуха. Продукт получают в виде масла (1 О 2,5 г).1 Аллиловьй зфир стадии с(1 ОО 5 глоту и экстрагируют эфиром, который промывают разбавленной соляной кислотой и затем водой. Органический раствор экстрагируют 10 ным раствором гидроокиси натрия н затем 5 подкисляют.0 сажденны продукт экстрагируют эфиром, которьй сушат над сульфатом магния. Полученный эфирнй раствор упаривают досуха с получением желтоткорнчневого маслаЭту смесь растворяют в этаноле(20 мл) и гидрируют в присутствии катализатора Адамса до тех пор,пока не прекратится принятие водорода. Катализатор отфилътровыеают через киэельгур, и фильтрат уваривают до коричневого масла 179,9 г). Коричневое масло является смесью и разделяд ется (при помощи хроматографии с жидкостью высокого давления, раствод рнтелъ система эфир/петролейный эфир 11) с получением д 42 г соединения этой стадии и 23,8 г 6-(ЫацетилЫэтил)амнноЗпропнл 2 оксиацетофенона. 04-(НАцетилЫэтил)амнноЗпро- 35 пил 2 оксиацетофенон (ад г) нагревают с обратным холодильником В 482 ном бромистом водороде в ледяной уксусной кислоте (100 мл), ледяной уксусной кислоте (500 мл) и воде(20 мл) в течение 6 ч. Реакционную смесь сливают на ледяную воду И Экстрагируют этилацетатом, который промывают водой, раствором бикарбоната натрия, затем опять водой и сушат-над сулфатом магния. Органический растворителътупаривают ДОСУХЗ С ПО лучением продукта этой стадии ввиде красного масла (34 г). СтрУКТУ ра подтверждена ЯМР и массспектроскопией.. Амин стадии Ь(17 г) и дшетнлацетилендикарбоксилат (113 мл) На гревают в этаноле (300 мл) С Обрат ны колодилъником в течение 17 Ч 40Реакционную смесь охлаждают и унарнвают досуха с получением яркокрас ного масла. Это масло подвергают хроматографии на силикагеле на колонке с использованием системы эфир/петролейный эфир 11 в качестве раэБа вителя с получением 19,1 г диметнл(100 мл) на паровой бане в течение 10 мин. Реакционную смесь охлаждают и слиают на смесь ледяной воды и этилацетата. Органически раствор отделяют, промывают водой нсушат над сульфатом магния. Растворитель упариъают досуха с получением бледное желтого твердого вещества Этот прогдукт онищают при помощи цидкрй кр Й матографии-высокогодавления с надула чеиием 2,6 г соединенияэтой стадии, т.лл. 121123 С. . о Найдено,2 С 65,5 Н 6,6 Ы,д,2. с 1 вн 21 Н 05 Вычислено, С 65,3 Н 6,34 П 4,23. 5При очистке бледножелтого твердого вещества получают метилт 6 адетилОксикетон стадии 6(1,О г) и дн этилоксалат (33 мл) в ЦУХОМ дИМеТНЛ формамнде (25 мл) добавляют в эфир,промтый 502 нм гидридом натрия.20 мл), н реакционную смесь перемешивают 4 ч. Реакционную смесь сливают,этилацетатом,ксторый затемпромывдют водой и сушат над сульфатом магния. Растворитель упаривают досуха с ПОЛУченнем масла, которое растворено В этаноле (100 мл),и добавляют концентрированнуюсоляную кислоту (несколь-ко капель). Раствор нагревают С 05 ратным холодильником в течение 0,5 ч,охлаждают, сливаютв воду и экстрагируют этилацетатом, который ПРОМНВаЮТ водой и сушат над сУлъФаТ 0 М магнияРастворитель упариают ДОСУКЗ С ПО лучением масла, которое отверждается при растирании с д 060-иым петролейным эфиром (12 г). Структура Ч 0 едИ нения подтверждается ЯМРспектР 0 М 7 1152523 - 8(85 мл) и воде (32 мл) нагревают в течение 4 ч. Реакционную смесь слит вают в водУ подкисляют,-осадок собирают фильтрацией И сушат. родукт очищают при помощи растирания в порошок с кипящим этанолом, затем дважды с кипящим ацетоном. После каждого растирания смесь центрифу 10гируют И всплывающую жидкость удаляют декантацией. Оставшееся твердое 15вещество сушат с получением 0,547 г требуемой дн-кислоты в виде желтого порошка, тпл. 2983 ООС(раэл.). Найдено,2 С 61,3 Н 5,0 Ы 3,6. СНЫО 7 ч. Вычислено 2 С 61,53 Н 4,6 Ы 3,79, 4 2. Динатрнй 46 диоксо 1 этнлТОпропил 4 Н 6 Нпирано(З,2-)хинопин 28 димарбоксилат. Указанную днкислоту (4,098 г),суспендированную в воде (100 Мл),обрабатывают бикарбонатом натрия 11,82 г). Полученный раствор фильтруют, и фильтрат обрабатывают ацетоном до тек пор, пока не произойдет полного осаждения продукта. Требуемую динатрневую соль ФНЛБТРУЮТ. и сушат с получением 3,39 г бледножелтого порошка. найдено С 51,1 Н 4,3 Ы 3.07 СэН 5 ЫЫа 1069 НдОВычислено С 51,1 В 4,1 Ы 31.Пр и ме р 3. Предлагаенм способом могут быть получены следующие соединения 1. 5 Этилч 48 дноксо 10 пропил 4 НВНпирано(2,ЗЬ)кинолин-2,6 дикарбоновая кислота.П ри м е р 4 Предлагаемым способом могут быть получены также следуюие-соединенияП рки ме р 5. Предлагаемым спобом Могут быть получены также следующие соединенияЪ. 46-Диоксо-10-(проп 2 енил) 4 Н 6 Нпирано(3,2-е)хинолин 2,8 ди карбоновая кислота структура под 7 тверждена ЯМРтспектромь
МПК / Метки
МПК: C07D 491/052, A61K 31/47
Метки: солей, или, получения, приемлемых, пиранохинолинов, способ, фармацевтически
Код ссылки
<a href="https://by.patents.su/8-164-sposob-polucheniya-piranohinolinov-ili-ih-farmacevticheski-priemlemyh-solejj.html" rel="bookmark" title="База патентов Беларуси">Способ получения пиранохинолинов или их фармацевтически приемлемых солей</a>
Способ получения осветленной крови убойных животных

Номер патента: 22
Опубликовано: 30.05.1994
Авторы: Романюк Е. Н., Полунченко В. В., Фадеева Л. Я., Варганов В. А.
МПК: A23L 1/06
Метки: получения, убойных, крови, осветленной, животных, способ
Текст:
...к массе стабилизированной крови позволяют достигнутьпоставленнуюцель. т Повышение доли крови неизбежно ска ЗЫВЭВТСЯ на ООГЭНОЛЕПТИЧВСКИХ ПОКЗЗЭТЕляхпродукта. он приобретает более темную окраску. Повышение доли обезжиренногоСГУЩВННОГО МОЛОКЗ ПрИВОДИТ К СНИЖЕНИЮсгущенным молоком приводит к перераспт уределению частиц белков крови и молока.Уксуснаят кислота используется в качестве катализатора в этом процессе. В результате получается однородная...
Способ получения сухого продукта из молочной сыворотки

Номер патента: 21
Опубликовано: 30.05.1994
Авторы: Романюк Е. Н., Иванов А. Т., Фадеева Л. Я., Варганов В. А., Галушко В. М., Полунченко В. В., Гирис Д. А.
МПК: A23C 21/00
Метки: способ, молочной, сухого, получения, сыворотки, продукта
Текст:
...состав сухого продукта по сравнению с сухим молочным продуктом.Увеличение содержания белка и снижение содержания лактозы способствует УСТЮЭНЕНИЮ НЭПИПЭНИЯ продукта на нут 10ранних поверхностях сушильной установки,улучшению сыпучих свойств сухого продукТОЕ. СНИЖЕНИЮ ЕГО ГИГПОСКОПИЧНОСТИ.При СУШКЕ СМЕСИ С СОСТНОШЕНИЕМ 80 мас. 34. сухих веществ сыворотки и 20 масд, сухих веществ крови оптимальными являются следующие параметры сушки 18 ОС на...
Способ получения битумной эмульсионной мастики

Номер патента: 42
Опубликовано: 30.05.1994
Авторы: Маслаков Аркадий Дмитриевич, Вилисов А. П.
МПК: C08L 95/00
Метки: битумной, эмульсионной, получения, способ, мастики
Текст:
...шхту зчтльгатора при непрешшщмпфшшшшышавтщшше10 мин поочередно (порционно за д раза) вводят предварительно разогре тьш до 1 ос битум (маркиБНД 60/90)(47 мас.) н оставшееся количество 5 воды (333 мас.). П р н м е р 2 Битумную мастику готовят как в примере 1, но при этом для получения шихты (суспензии) эмульгатора берут 5 мас.2 извести негашеной, 13 мас.2 асбеста и 3,35 мас.2 воды (10 мас. от общего иоличества воды), при этом битум...
Способ выделения катализатора на основе ацетатов кобальта и марганца из остатка производства диметилтерефталата

Номер патента: 113
Опубликовано: 30.09.1994
Авторы: Герхарт Гоффман, Рудольф Кордес, Гейнрих Бюнгер
МПК: B01J 31/40
Метки: основе, ацетатов, способ, выделения, марганца, производства, диметилтерефталата, кобальта, остатка, катализатора
Текст:
...20 ч. Обе фазы непрерывно отводят из отстойника 6, причем органическую фазу подают в отстойник 4 при помощи лопастного насоса 10, во всасыающую трубу 9 которого одновременно с органической фазой подают 210 кг/ч реакционной воды. при этом соотношение органической фазы и реакционной воды, служащей в качестве экстрагента, составляет 10,27, а им среднее время пребывания в лопастном насоса 10 равно 1,2 с. Получаемую в насосе смесь подают в...
Предыдущий патент: Устройство для выявления сердечной аритмии
Следующий патент: Устройство для экстрагирования твердых веществ жидкостью
Случайный патент: Дифференциал повышенного трения (варианты)