Модифицированные углеводами цитостатические средства
Номер патента: 5307
Опубликовано: 30.06.2003
Авторы: Уве ПЕТЕРЗЕН, Михаэль ШПЕРЦЕЛЬ, Клаус-Дитер Бремм, Карстен ФОН ДЕМ БРУХ, Вальтер ВАЙХЕЛЬ, Ханс-Георг ЛЕРХЕН, Норберт ПИЛЬ, Хорст-Петер АНТОНИЦЕК, Ерг БАУМГАРТЕН




Текст
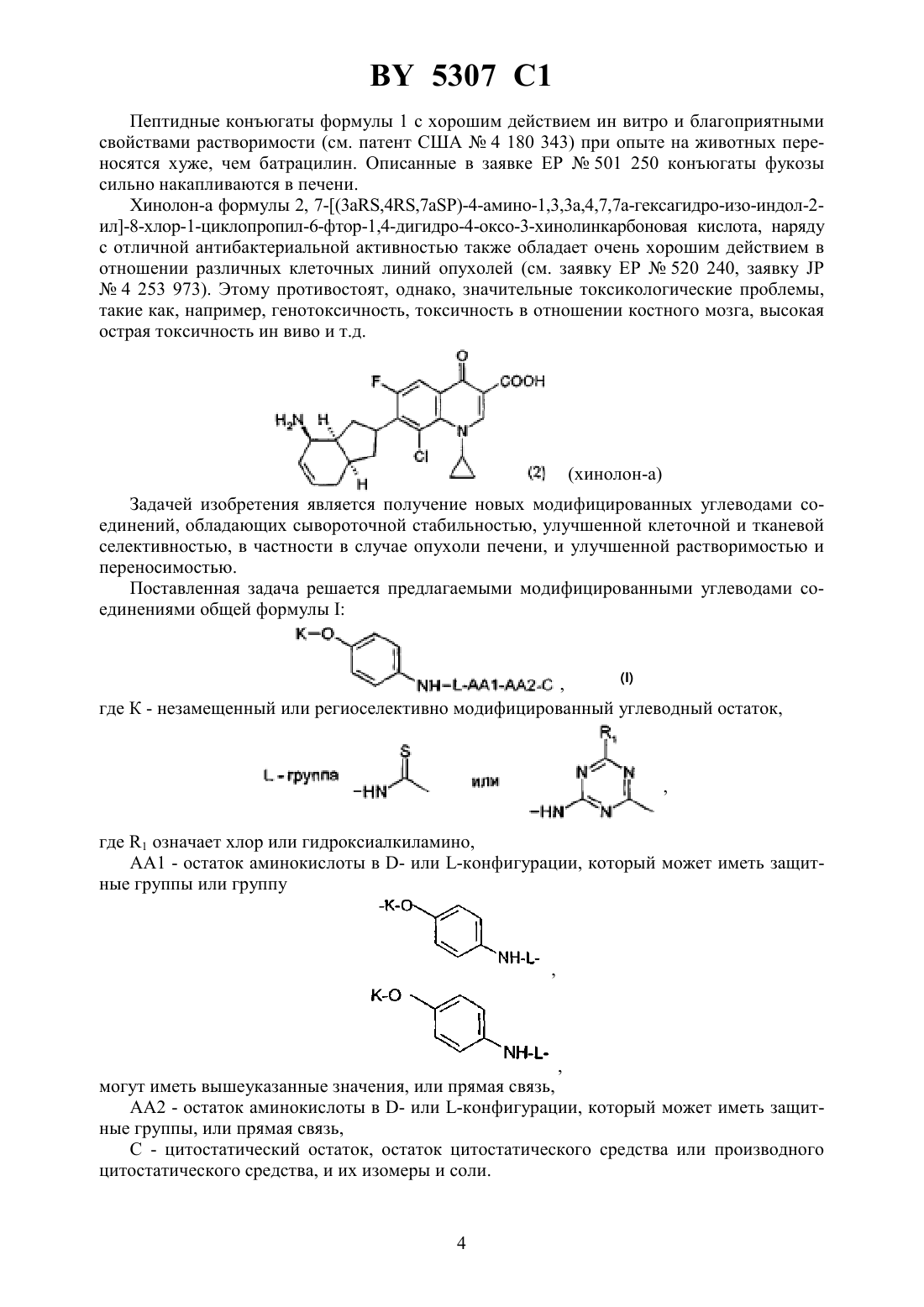
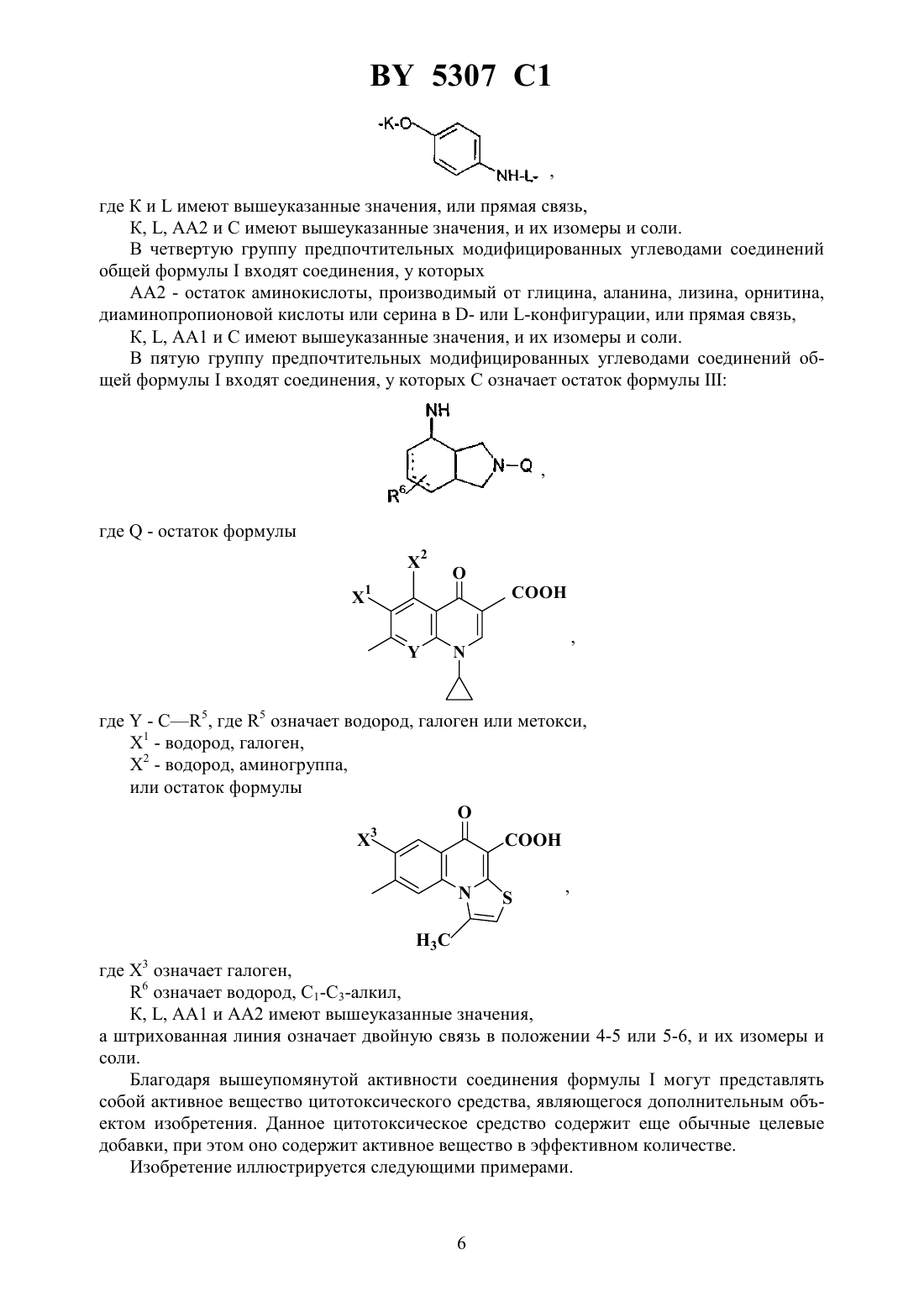
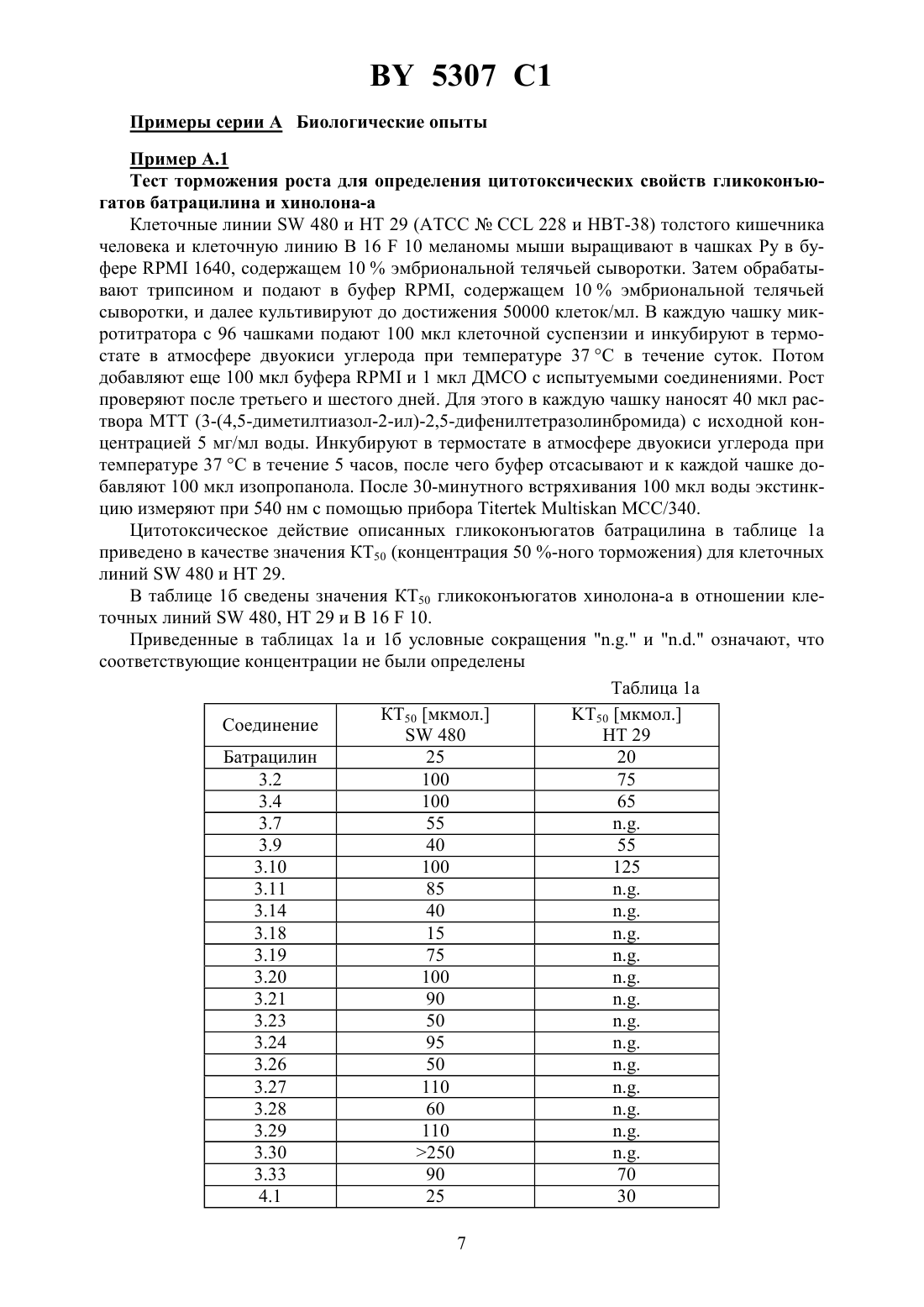
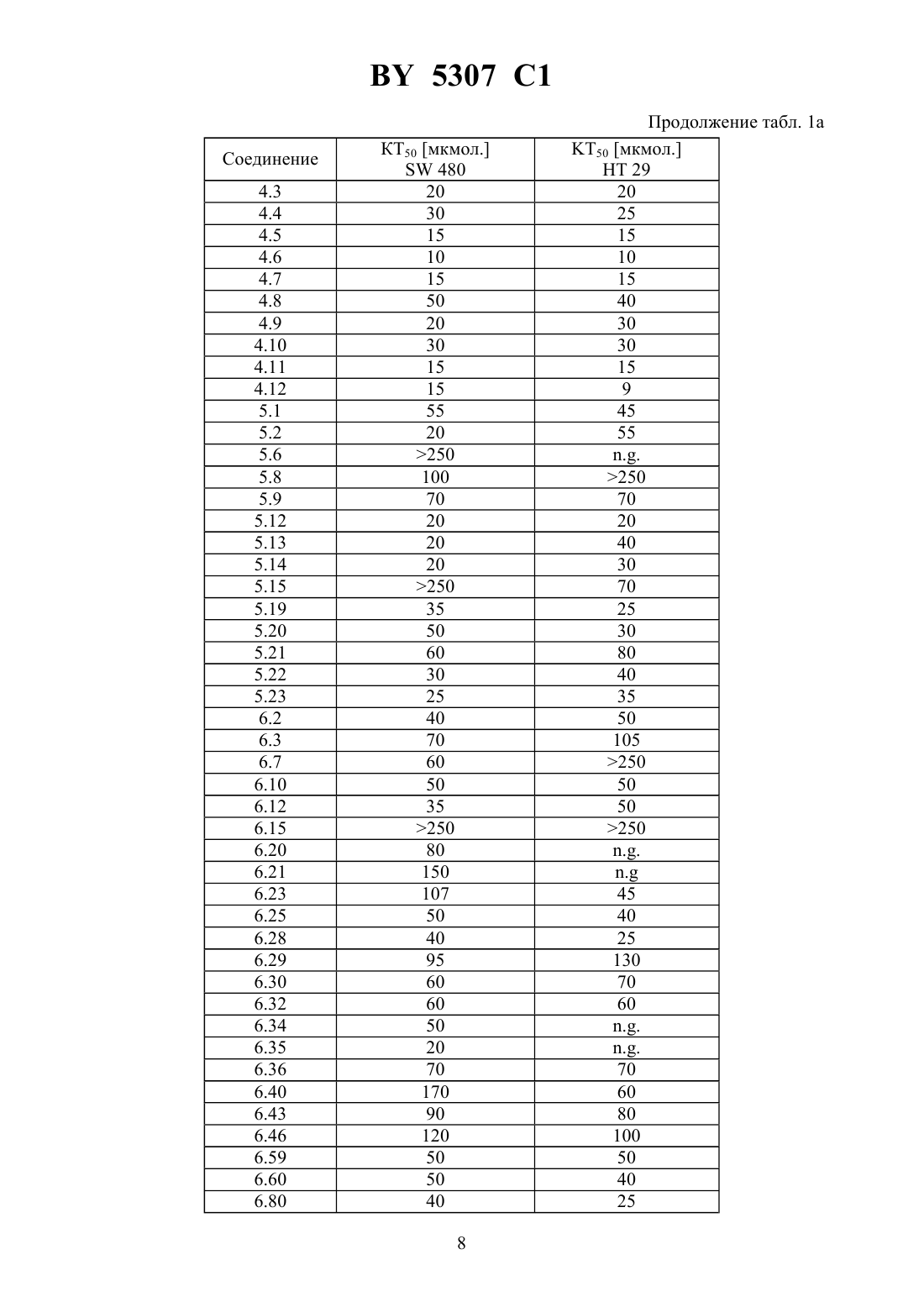
НАЦИОНАЛЬНЫЙ ЦЕНТР ИНТЕЛЛЕКТУАЛЬНОЙ СОБСТВЕННОСТИ МОДИФИЦИРОВАННЫЕ УГЛЕВОДАМИ ЦИТОСТАТИЧЕСКИЕ СРЕДСТВА(72) Авторы Ханс-Георг ЛЕРХЕН Карстен ФОН ДЕМ БРУХ Уве ПЕТЕРЗЕН Ерг БАУМГАРТЕН Норберт ПИЛЬ Хорст-Петер АНТОНИЦЕК Вальтер ВАЙХЕЛЬ Михаэль ШПЕРЦЕЛЬ Клаус Дитер Бремм(57) 1. Модифицированные углеводами соединения общей формулы где К - незамещенный или региоселективно модифицированный углеводный остаток,1 где 1 - хлор или гидрокси-С 1-С 6-алкиламино,АА 1 - остаток аминокислоты в - или -конфигурации, который может иметь защитные группы или группу,в которойинезависимо оти , содержащихся в формулев группе,могут иметь вышеуказанные значения, или прямая связь,АА 2 остаток аминокислоты в - или -конфигурации, который может иметь защитные группы, или прямая связь,Сцитостатический остаток, выбранный из остатка цитостатического средства и остатка производного цитостатического средства,его изомеры или соли. 2. Модифицированное углеводами соединение по п. 1, отличающееся тем, что ,1, 2 иимеют указанные в п. 1 значения, Куглеводный остаток общей формулы где А - метил, гидроксиметил, С 1-С 6-алкоксиметил или карбокси-С 1-С 6-алкилоксиметил,2, 3 и 4 отдельно или вместе означают водород, гидрокси, С 1-С 6-алкилокси, гидрокси-С 1-С 6-алкилокси, карбокси-С 1-С 6-алкилкарбонилокси, сульфат или галоген, или два из 2, 3 и 4 вместе означают эпоксигруппу,его изомеры или соли. 3. Модифицированное углеводами соединение по п. 1, отличающееся тем, что К, ,1 и 2 имеют указанные в п. 1 значения,- производное хинолонкарбоновой кислоты(а, б, в или г), батрацилин, камптотецин, доксорубицин или мелфалан, его изомеры или соли. 4. Модифицированное углеводами соединение по п. 1, отличающееся тем, что К, ,2 иимеют указанные в п. 1 значения, 1 остаток аминокислоты, выбранный из глицина, аланина, валина, лизина, аспарагиновой кислоты, глутаминовой кислоты, орнитина, тирозина и серина, в - или -конфигурации, который может быть замещен группой где К иимеют указанные в п. 1 значения, или прямая связь, его изомеры или соли. 5. Модифицированное углеводами соединение по п. 1, отличающееся тем, что К, ,1 иимеют указанные в п. 1 значения, 2 - остаток аминокислоты, выбранной из глицина, аланина, лизина, орнитина, диаминопропионовой кислоты и серина, в - или конфигурации, или прямая связь, его изомеры или соли. 6. Модифицированное углеводами соединение по п. 1, отличающееся тем, что К, ,1 и 2 имеют указанные в п. 1 значения,остаток общей формулы где- остаток общей формулы 2 где- -5, где 5 водород, галоген или метокси,1 - водород или галоген,2 - водород, амино или остаток общей формулы 5307 1 пунктирная линия означает двойную связь в положении 4-5 или 5-6,его изомеры или соли. 7. Цитостатическое средство, содержащее активное вещество и целевые добавки, отличающееся тем, что в качестве активного вещества содержит соединение по любому из пп. 1-6 в эффективном количестве. Изобретение относится к новым цитостатическим средствам, специфичным к опухолям, в частности к модифицированным углеводами соединениям цитостатической активности. Химиотерапия при опухолевых заболеваниях сопровождается в большинстве случаев серьезными побочными действиями, обусловленными токсичностью химиотерапевтических средств в отношении пролиферирующих клеток других тканей. Уже много лет ученые занимаются проблемой улучшения селективности применяемых активных начал. Интенсивно разрабатывается синтез пролекарств, которые более или менее селективно высвобождаются в целевой ткани, например, в результате изменения значения рН (см.и др., например, патент 4 229 903), с помощью ферментов (например, глюкуронидаз см.и др., заявку ЕР 511 917,и др., заявку ЕР 595 133) или антитело-ферментных конъюгатов (см.и др., заявку 88/07378,и др., патент США 4 975 278,и др., заявку ЕР 595 133). Проблематичными при этом оказываются, среди прочего, недостаточная стабильность конъюгатов в других тканях и органах и, в частности, повсеместное распределение активного начала, происходящее после внеклеточного высвобождения активного начала в опухолевой ткани. Выраженная структура лектинов на поверхности опухолевых клеток (см.в источнике 12, 1989 г., стр. 175) предоставляет принципиальную возможность путем связывания соответствующих компонентов углеводов с цитостатическими средствами целенаправленно воздействовать на опухолевые клетки. Эта перспектива ограничивается тем, что и в других тканях, в частности в печени, имеются лектины сходной специфичности в отношении углеводов (например галактозы, лактозы, маннозы, -ацетил-глюкозамина,фукозы и т.д.) (см.и др. в источнике . . . 46, 1982 г., стр. 531 и др. в источнике . . . .74, 1977 г., стр. 1521 и др. в источнике . . . 262, 1986 г., стр.7433 и др. в источнике . . . 266, 1991 г.,стр. 3343). Поэтому следует ожидать значительного накопления содержащих активное начало гликоконъюгатов в печени и в других богатых лектинами органах, если применяют такие немодифицированные сахара. Гетероциклический амин батрацилин формулы 1 проявляет хорошее противоопухолевое действие в различных моделях рака кишки (см. патент США 4 757 072). 5307 1 Пептидные конъюгаты формулы 1 с хорошим действием ин витро и благоприятными свойствами растворимости (см. патент США 4 180 343) при опыте на животных переносятся хуже, чем батрацилин. Описанные в заявке ЕР 501 250 конъюгаты фукозы сильно накапливаются в печени. Хинолон-а формулы 2, 7-(3,4,7)-4-мино-1,3,3,4,7,7-гексагидро-изо-индол-2 ил-8-хлор-1-циклопропил-6-фтор-1,4-дигидро-4-оксо-3-хинолинкарбоновая кислота, наряду с отличной антибактериальной активностью также обладает очень хорошим действием в отношении различных клеточных линий опухолей (см. заявку ЕР 520 240, заявку 4 253 973). Этому противостоят, однако, значительные токсикологические проблемы,такие как, например, генотоксичность, токсичность в отношении костного мозга, высокая острая токсичность ин виво и т.д.(хинолон-а) Задачей изобретения является получение новых модифицированных углеводами соединений, обладающих сывороточной стабильностью, улучшенной клеточной и тканевой селективностью, в частности в случае опухоли печени, и улучшенной растворимостью и переносимостью. Поставленная задача решается предлагаемыми модифицированными углеводами соединениями общей формулы,где К - незамещенный или региоселективно модифицированный углеводный остаток,,где 1 означает хлор или гидроксиалкиламино,АА 1 - остаток аминокислоты в - или -конфигурации, который может иметь защитные группы или группу,могут иметь вышеуказанные значения, или прямая связь,АА 2 - остаток аминокислоты в - или -конфигурации, который может иметь защитные группы, или прямая связь,С - цитостатический остаток, остаток цитостатического средства или производного цитостатического средства, и их изомеры и соли. в целом представляет собой спейсер, связывающий К и С. Соединения формулымогут иметься в стереоизомерных формах, например в виде энантиомеров или диастереомеров, или в виде их смесей, например в виде рацемата. Изобретение относится к чистым стереоизомерам, а также их смесям. В случае необходимости смеси стереоизомеров можно разделять известным образом на единые стереоизомерные компоненты, например путем хроматографии или способов кристаллизации. Соединения формулымогут также иметься в виде их солей. В общем, здесь можно назвать соли с органическими или неорганическими основаниями или кислотами, а также внутренние соли. В число добавляемых кислот входят предпочтительно галогенированные водородные кислоты, например хлористоводородная кислота и бромистоводородная кислота, в частности хлористоводородная кислота, далее фосфорная кислота, азотная кислота, серная кислота, моно- и бифункциональные карбоновые кислоты и гидроксикарбоновые кислоты,такие как, например, уксусная кислота, малеиновая кислота, малоновая кислота, оксалевая кислота, глюконовая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, салициловая кислота, сорбиновая кислота и молочная кислота, а также сульфокислоты, такие как, например, п-толуолсульфокислота, 1,5-нафталиндисульфокислота или камфарная сульфокислота. Физиологически невредные соли могут представлять собой металлические или аммониевые соли предлагаемых соединений, имеющие свободную карбоксильную группу. Особенно предпочтительными являются, например, соли натрия, калия, магния или кальция,а также аммониевые соли, которые производятся от аммиака или органических аминов,таких как, например, этиламин, ди- или триэтиламин, ди- или триэтиноламин, дициклогексиламин, диметиламиноэтанол, аргинин, лизин, этилендиамин или фенэтиламин. В первую группу предпочтительных модифицированных углеводами соединений общей формулывходят соединения, у которых К - углеводный остаток формулы где А - метил, гидроксиметил, алкоксиметил, карбоксиалкилоксиметил,2, 3, 4 отдельно или вместе означают водород, гидроксил, алкилокси, гидроксиалкилокси, карбоксиалкилкарбонилокси, сульфат, или два из остатков 2, 3, 4 вместе означают эпоксигруппу 1, АА 2 и С имеют вышеуказанные значения, и их изомеры и соли. Во вторую группу предпочтительных модифицированных углеводами соединений общей формулывходят соединения, у которых С - хинолонкарбоновая кислота (а, б, в и г),батрацилин, камптотецин, доксорубицин, мелфалан,К, , 1 и АА 2 имеют вышеуказанные значения, и их изомеры и соли. В третью группу предпочтительных модифицированных углеводами соединений общей формулывходят соединения, у которых АА 1 - остаток аминокислоты, производимый от глицина, аланина, валина, лизина, аспарагиновой кислоты, глутаминовой кислоты,орнитина, тирозина, или серина в - или -конфигурации, который может быть связан с дополнительной группой 5,где К иимеют вышеуказанные значения, или прямая связь,К, , 2 и С имеют вышеуказанные значения, и их изомеры и соли. В четвертую группу предпочтительных модифицированных углеводами соединений общей формулывходят соединения, у которых АА 2 - остаток аминокислоты, производимый от глицина, аланина, лизина, орнитина,диаминопропионовой кислоты или серина в - или -конфигурации, или прямая связь,К, , 1 и С имеют вышеуказанные значения, и их изомеры и соли. В пятую группу предпочтительных модифицированных углеводами соединений общей формулывходят соединения, у которых С означает остаток формулы где Х означает галоген,6 означает водород, С 1-С 3-алкил,К, , 1 и АА 2 имеют вышеуказанные значения,а штрихованная линия означает двойную связь в положении 4-5 или 5-6, и их изомеры и соли. Благодаря вышеупомянутой активности соединения формулымогут представлять собой активное вещество цитотоксического средства, являющегося дополнительным объектом изобретения. Данное цитотоксическое средство содержит еще обычные целевые добавки, при этом оно содержит активное вещество в эффективном количестве. Изобретение иллюстрируется следующими примерами. 5307 1 Примеры серии А Биологические опыты Пример А.1 Тест торможения роста для определения цитотоксических свойств гликоконъюгатов батрацилина и хинолона-а Клеточные линии 480 и НТ 29 (АТСС 228 и НВТ-38) толстого кишечника человека и клеточную линию В 1610 меланомы мыши выращивают в чашках Ру в буфере 1640, содержащем 10 эмбриональной телячьей сыворотки. Затем обрабатывают трипсином и подают в буфер , содержащем 10 эмбриональной телячьей сыворотки, и далее культивируют до достижения 50000 клеток/мл. В каждую чашку микротитратора с 96 чашками подают 100 мкл клеточной суспензии и инкубируют в термостате в атмосфере двуокиси углерода при температуре 37 С в течение суток. Потом добавляют еще 100 мкл буфераи 1 мкл ДМСО с испытуемыми соединениями. Рост проверяют после третьего и шестого дней. Для этого в каждую чашку наносят 40 мкл раствора МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолинбромида) с исходной концентрацией 5 мг/мл воды. Инкубируют в термостате в атмосфере двуокиси углерода при температуре 37 С в течение 5 часов, после чего буфер отсасывают и к каждой чашке добавляют 100 мкл изопропанола. После 30-минутного встряхивания 100 мкл воды экстинкцию измеряют при 540 нм с помощью прибора/340. Цитотоксическое действие описанных гликоконъюгатов батрацилина в таблице 1 а приведено в качестве значения КТ 50 (концентрация 50 -ного торможения) для клеточных линий 480 и НТ 29. В таблице 1 б сведены значения КТ 50 гликоконъюгатов хинолона-а в отношении клеточных линий 480, НТ 29 и В 1610. Приведенные в таблицах 1 а и 1 б условные сокращения и означают, что соответствующие концентрации не были определены Таблица 1 а КТ 50 мкмол. 50 мкмол. Соединение 480 НТ 29 Батрацилин 25 20 3.2 100 75 3.4 100 65 3.7 55 0,3 12.5 1 4 0,2 12.6 4 7 0,3 12.7 60 250 20 12.8 8 7 1 12.9 4 8 2 12.10 15 15 4 12.11 2 2 0,5 12.12 8 13 0,5 12.13 35 100 1 12.14 1 2 0,3 12.15 0,3 1 0,1 14.1 0,8 1 1,5 14.2 1 6 1,5 14.3 8 4 4 14.4 1,5 1 0,4 15.1 20 20 2 15.2 50 70 15 16.1 50 100 200 16.2 50 60 80 17.1 10 5 5 17.2 4 4 4 18.1 0,03 0,01 0,2 18.2 0,02 0,02 0,2 18.4 0,02 0,02 0,3 18.5 0,2 0,2 1 18.9 0,08 0,06 0,7 18.14 0,015 0,01 0,08 Зависимость биологического действия от углеводов дополнительно доказана неактивностью сравнительных соединений серий примеров 5, 6 и 11 -,-бис-(4-гидроксифениламино-тиокарбонил)-лизил-батрацилин и -,-бис-(4-гидроксифениламинотиокарбонил)-лизилаланил-батрацилин или -,-бис-(4-гидроксифениламинотиокарбонил)лизил-хинолон-а (значения КТ 50250). 9 5307 1 Пример А.2 Испытание ин витро на расщепляемость гликоконъюгатов Кинетика расщепления с применением крови человека 1,225 мл крови человека вместе с 1,25 мл фосфатно-солевого буферного раствора и 25 мкл маточного раствора субстрата (1 мг/мл в 3 ДМСО в фосфатно-солевом буферном растворе) инкубируют при температуре 37 С. Через 1 час и через 24 часа отбирают пробы в количестве по 1 мл, смешивают с 1 мл этанола, после чего оставляют стоять при 4 С в течение 20 минут. Центрифугируют 5 минут при 3500 об/мин, после чего отбирают 100 мкл надосадочной жидкости для анализа с помощью высокоскоростной колоночной жидкостной хроматографии. Кинетика расщепления с применением клеток 2,25 мл фосфатно-солевого буферного раствора вместе с 225 мкл клеточной суспензии(30 мг/мл) и 25 мкл маточного раствора субстрата (1 мг/мл в 3 ДМСО в фосфатносолевом буферном растворе) инкубируют при температуре 37 С. Через 1 час и через 24 часа отбирают пробы в количестве по 1 мл, смешивают с 1 мл этанола, после чего оставляют стоять при 4 С в течение 20 минут. Центрифугируют 5 минут при 3500 об/мин, после чего отбирают 100 мкл надосадочной жидкости для анализа с помощью высокоскоростной колоночной жидкостной хроматографии. Условия высокоскоростной колоночной жидкостной хроматографии Хроматограф прибор фирмыКолонна марки 18 5 мкм 2504 мм Элюент А 10 ммол. буфера фосфата калия, рН 4,5 Б смесь 80 ацетонитрила и 20 воды Скорость подачи 1 мл/мин Длина волны 372 нм Градиент 0 мин 10 Б 10 мин 60 Б 15 мин 60 Б 18 мин 10 Б 20 мин 60 Б Элюент конъюгатов хинолона-а А 100 метанола Б 10 ммол. буфера фосфата калия, рН 2,2 10 ммол. гептансульфокислоты Таблица 2 а Пример 3.4 3.9 4.4 5.9 6.2 6.12 7.3 Продуктом расщепления является аланил-батрацилин. 10 Пример А.3 Испытание по распределению в органах Во всех экспериментах были использованы атимические голые мыши штамма/, которых разводят в лаборатории,проф. Фибига в г. Фрайбурге. Животных держали в поликарбонатных клетках (торговой марки Макролон) в условиях ламинарного потока. В качестве опухолевого материала употребляли ткань клеточной линии 480, которая предварительно несколькими операциями переноса была введена в мыши. Голым мышам в возрасте 6-8 недель подкожно имплантировали в бока по двум опухолям на животное. До рандомизации животных держали в течение 26-27 суток. Тогда средний размер опухолей составлял 500 мг и радиус опухолей - около 10 мм. Сама фармакокинетика произошла следующим образом голым мышам инъецировали испытуемое соединение, после чего мышей хранили еще в клетках до отбора пробы через полчаса или 4 часа. Отбор пробы начался с взятия крови. Для этого мышь наркотизировали эфиром в течение 30-60 сек. Через 0,5 ч или же 4 ч после введения соединения открыли брюшную полость, мышь в наркозе обескровливали через венув течение 1-2 минут и затем умерщвляли путем перелома шейного отдела позвоночника. В результате этого происходила остановка системы кровообращения и прекращение перфузии органов. В дальнейшем отдельные органы освобождали и вынимали, что длилось около 5 минут. Непосредственно после этого взвесили пробы органов и затем остальное тело и замораживали в жидком азоте. Соединение конъюгат 1 внутрибрюшинно вводили в количестве 300 мг/кг веса тела,а соединение конъюгат 2 вводили в количестве 100 мг/кг в хвостовую вену. При проведении опыта для каждого соединения и каждой дозы были использованы по 5 животным. Результаты распределения конъюгата 1 приведены в таблице 3, а конъюгата 2 - в таблице 4. А. Калибровка 5, 10, 50, 100 и 200 мкг соединения, растворенного в смеси этанола и воды в объемном соотношении 11, добавляли к 1 г печени коровы. Затем измельчали с помощью 1 г песка и 2,5 мл охлажденной смеси этанола и воды в объемном соотношении 11 и пробы центрифугировали в течение 2 минут при 3500 об/мин. Надосадочную жидкость удаляли, остаток еще раз перемешивали с 2,5 мл смеси этанола и воды, центрифугировали и супернатанты объединяли. Отбирали по 100 мкл и подвергали анализу с помощью высокоскоростной колоночной жидкостной хроматографии. Условия высокоскоростной колоночной жидкостной хроматографии Хроматограф прибор фирмыКолонна марки 18 5 мкм 2504 мм 11 Скорость подачи Длина волны Б. Обработка органов Обработку осуществляли аналогично А, причем все органы обрабатывали 2,5 мл смеси этанола и воды и экстрагировали. В. Обработка крови Отобранное количество крови перемешивали с 2 мл смеси этанола и воды в объемном соотношении 11, центрифугировали в течение 2 минут при 3500 об/мин и надосадочную жидкость декантировали. К остатку еще раз добавляли 2 мл смеси этанола и воды в объемном соотношении 11, центрифугировали и супернатанты объединяли. Отбирали по 100 мкл объединенных супернатантов и подвергали анализу с помощью высокоскоростной колоночной жидкостной хроматографии в условиях, приведенных под п. А. Конъюгат 1 (заявка ЕР 501 250) 5307 1 Таблица 3 Орган Кровь Опухоль Печень Почка Мозг Анализ проб органов Средняя величина после 1 ч (мкг/г органа) 31,2 56,6 770 34,1 81 26,1 Средняя величина после 4 ч (мкг/г органа) 24,4 126 22,4 78,4 27,5 Таблица 4 Орган Кровь Опухоль Печень Почка Мозг Анализ проб органов Соединение Средняя величина после 0,5 ч (мкг/г органа) Конъюгат 2 6,5 Батрацилин Конъюгат 2 2,0 Батрацилин 2,5 Конъюгат 2 0,9 Батрацилин Конъюгат 2 17,5 Батрацилин 10,7 Конъюгат 2 Батрацилин Средняя величина после 4 ч (мкг/г органа) 3,12 1,2 0,5 0,8 Пример А.4 Определение острой токсичности гликоконъюгатов батрацилина (разовое введение) Острую токсичность производных батрацилина определяли на голых мышах штамма/. Внутривенно введенные соединения один раз инъецировали мышам в виде водных растворов, а внутрибрюшинно введенные соединения - в виде растворов ДМСО в концентрациях до 400 мг соединения на кг мыши. Переносимую разовую дозу вычисляли на основе потери веса животных до 21-го дня после введения соединения и на основе числа выживших животных. Переносимые разовые дозы соединений приведены в таблице 5. В случае соединений примеров 3.16, 3.33, 3.9, 6.12, 6.14, 6.2, 6.81, 8.2 доза составляла свыше 200 мг соединения на кг мыши. В случае соединений примеров 3.33, 6.12, 6.14, 6.2,6.81 даже при дозе 400 мг/кг острой токсичности не было найдено. В отличие от этого не содержащий сахара лизилаланил-батрацилин примера 2.13 проявлял заметную токсичность уже при разовых дозах, составлявших от 25 до 50 мг на кг мыши. 13 5307 1 Таблица 5 Максимально переносимая разовая доза производных батрацилина и производных хинолона-а Пример Переносимая разовая доза в мг/кг мыши Внутрибрюшинное введение Внутривенное введение 2.13 50 25 3.4 200100 (2 животных погибли) 3.9 200 200 3.16200 не опред. 3.33 не опред.400 6.2400 не опред. 6.12 не опред.400 6.14400 не опред. 6.81 не опред.400 8.2200 не опред. Хинолон-а не опред.25 12.3 не опред.200 Пример А.5 Определение острой токсичности после многократного введения Часть соединений ввели внутривенно, другую часть - внутрибрюшинно, либо ежедневно в дни с 1-4 и с 7-10, либо в дни 1, 5 и 9. Дозировки составляли 400, 200 и 100 мг/кг в день. Анализ осуществляли на основе потери веса животных до 21-го дня и на основе числа выживших животных. Для проведения опыта для каждого соединения и каждой дозы были использованы по 5 животным. Результаты приведены в таблице 6. Таблица 6 Максимально переносимая разовая доза при многократном введении Пример Пример А.6 Кроветворная активность гликоконъюгатов хинолона-а по сравнению с соответствующим активным началом Материал и методы Испытание ин витро Клетки костного мозга вымывали из бедра мыши. 105 клеток в среде марки 5(-) (торговый продукт Гензиме образование колоний основных клеток) и соеди 14 5307 1 нениями (10-4-100 мкг/мл) инкубировали при температуре 37 С в атмосфере, содержащей 7 двуокиси углерода. Спустя 7 дней определили число колоний ( 50 клеток) и кластеров (17-50 клеток). Испытание ин виво Мышам подкожно ввели 1, 3, 10, 30 мг/кг соединений. В различные промежутки времени (через 3, 24, 48, 72 ч. после инъецирования) удаляли бедра и выделяли клетки костного мозга. 2105 клеток вышеописанным образом инкубировали вместе с рекомбинантным мышиным колоние-стимулирующим фактором (-) и спустя 7 дней определили число колоний и кластеров. Результаты По данным таблицы 7 испытанные гликоконъюгаты по сравнению с хинолоном-а проявляли уменьшенное на фактор 105-103 торможение пролиферации основных клеток костного мозга. И при испытании ин виво по сравнению с хинолоном-а торможения пролиферации основных клеток соединением примера 12.3 в дозировке до 30 мг/кг не наблюдалось. Уже при введении 3 мг/кг хинолона-а было индицировано массивное подавление пролиферации основных клеток (см. рис. 1). Таблица 7 Индуцированная колоние-стимулирующим фактором пролиферация основных клеток костного мозга мыши Примеры Хинолон-а 11.2 11.7 12.1 12.3 12.6 12.8 14.1 14.2 14.4 Пример А.7 Противоопухолевая активность конъюгатов хинолона-а Активность ин витро гликоконъюгатов хинолона-а определяли на опухолевых ксенотрансплантатах человека в системе двухслойного культивирования на мягком агаре по Гамбургеру и Салмону (см. источник 197, стр. 461-463). Твердые опухоли сначала выращивали в атимической голой мыши штамма/, выделяли операцией и раздробляли механическим способом. Отдельные клетки получали потом в результате инкубации при 37 С в течение 30 минут в смеси ферментов, а именно 0,05 коллагеназы, 0,07 ДНКазы и 0,1 гиалуронидазы в буфере . Клетки два раза промывали, после чего их просеивали через сито с размером отверстий 200 мкм и 20 мкм. Применяли следующий способ культивирования Нижний слой содержал 0,2 мл модифицированной по способу Дульбекко среды Искова с 20 эмбриональной телячьей сыворотки и 0,7 агара. На этот слой наносили 40000200000 клеток в 0,2 мл той же самой среды и 0,4 агара в чашки микротитратора с 24 чашками. Цитостатические соединения добавляли в 0,2 мл среды. Культуры инкубировали в атмосфере, содержащей 7 двуокиси углерода, при температуре 37 С в течение 6-15 дней. Потом выросшие колонии подвергали количественному 15 5307 1 анализу в инвертированном микроскопе, при этом за 24 часа до анализа живущих колоний подкрашивали красителем на основе хлористого тетразола. Активность активного начала выражается ввыживших колоний по сравнению с числом колоний необработанных чашек (Т/Счисло колоний обработанных чашек 100/число колоний необработанных чашек). Соединение является активным, если значение Т/С 30 . В таблице 8 это значение приведено в качестве значения КТ 70 в мкг/мл. Таблица 8 Пример 280 НТ 294805295389898991023 Пример А.8 Испытания ин виво Метод В день 0 мышам инокулируют 5106 опухолевых клеток типа 1610. У животных развиваются твердые брюшинные опухоли, после чего им ежедневно вводят испытуемые соединения или же только носитель для них. В контрольной группе обычно погибает 50 животных в период с 14 по 20 день. Испытуемые соединения вводят в буфере или в системе органических растворителей, состоящей из 20 метанола и 20 диметилсульфоксида в 0,7 -ном растворе поваренной соли. Дача носителя не влияет на срок выживания животных. Терапевтический успех вытекает из продления времени выживания обработанных животных. Переносимость соединений анализируют параллельно на животных, не имеющих опухолей. По результатам переносимости и продления срока выживания можно ориентировочно определить терапевтический индекс. Таблица 9 выживших животных Контроль 11.12 1 мг/кг 11.12 100 мг/кг Хинолон-а 0,1 мг/кг Этопозид 5 мг/кг Таблица 9 показывает терапевтическую активность соединения примера 11.12 на мышах, которым трансплантировали опухоль типа 1610. 16 5307 1 Пример А.9 Торможение ин виво роста опухоля при применении модели голой мыши Материал Для всех экспериментов ин виво для испытания торможения роста опухоли использовали атимических голых мышей штамма/. Выбранный крупноклеточный рак легкого типа 529 выращивали в голых мышах серийными переносами. Человеческое происхождение опухоли было доказано изоферментативными и иммуногистохимическими методами. Проведение эксперимента Голым мышам штамма / в возрасте 6-8 недель опухоль подкожно имплантировали в оба бока. В зависимости от времени роста с удвоением объема лечение начали как только радиус опухолей достиг 5-7 мм. Мышей распределяли путем рандомизации по группе лечения и контрольной группе (5 мышей в каждой группе с 8-10 подвергающимися анализу опухолями). Все отдельные опухоли контрольной группы росли прогрессивно. Размеры опухолей измеряли с помощью раздвижного калибра в двух измерениях. Объем опухоли, который находится в хорошем соотношении с числом клеток, употребляли затем для всех анализов. Объем определяли по формуле длинаширинаширина /2(2/2, а илиозначают два прямоугольно расположенных диаметра). Значения относительного объема опухоли каждой отдельной опухоли определяли путем деления размера опухоли в день Н на размер опухоли в день 0 (в момент рандомизации). Средние значения относительного объема опухоли употребляли для дальнейшего анализа. Торможение увеличения объема опухоли (объем опухолей в испытуемой группе /контрольной группе, Т/С, в ) представляло собой окончательную измеренную величину. Лечение Все соединения давали по интермиттирующему плану соответственно в день 1, 5 и 9. Все соединения вводили внутрибрюшинно при использовании воды в качестве растворителя. Таблица 10 Терапия Доза а) Время выживания Число мг/кг/день число суток опухолей Относительный объем опухолидня 0 б) максимально переносимая доза в день 19 Соединения камптотецина в данном опыте проявляли, как правило, сравнимую или лучшую активность. б) 5307 1 Примеры серии Б Примеры получения соединений Пример 1.1 п-аминофенил-2 метилфукозид Раствор 750 мг (2,63 ммоль) п-нитрофенилфукозида в 40 мл смеси диметилформамида и диоксана в соотношении 12 при 0 С смешивают с 65 мг п-толуолсульфокислоты и 5 раз через каждые 30 минут 100 мкл 2-метоксипропена. После перемешивания при 20 С в течение 16 часов сгущают и очищают путем флеш-хроматографии с применением в качестве элюента смеси дихлорметана и метанола в соотношении 991. После сгущения получают 710 мг (83 ) белого твердого вещества. 1.1.б) п-нитрофенил-2 метил-3,4 изопропилиденфукозид 100 мг (0,307 ммоль) соединения примера 1.1.а) смешивают с 96 мкл метилйодида в 10 мл тетрагидрофурана, после чего порциями смешивают с 11 мг 80 -ного гидрида натрия. После перемешивания при 20 С в течение 3 часов к смеси добавляют еще 96 мкл метилйодида и 11 мг гидрида натрия. После дальнейшего перемешивания при 20 С в течение 16 часов к смеси добавляют небольшое количество воды и 100 мл дихлорметана. Реакционную смесь дважды встряхивают вместе с водой, органическую фазу сгущают и продукт очищают путем колоночной хроматографии с применением в качестве элюента смеси петролейного эфира и этилацетата в соотношении 81. Выход 78 мг (75 ). 1.1) п-аминофенил-2 метилфукозид 78 мг (0,23 ммоль) п-нитрофенил-2 метил-3,4 изопропилиденфукозида перемешивают при 20 С в течение 16 часов в 3 мл 80 -ной уксусной кислоты. Потом уксусную кислоту удаляют в вакууме, реакционную смесь смешивают с 10 мл метанола и после добавки двуокиси платины гидрируют в атмосфере водорода при незначительном избыточном давлении. Суспензию фильтруют на силикате марки , после чего фильтруемый материал промывают метанолом. После очистки путем хроматографии с применением в качестве элюента смеси дихлорметана и метанола в соотношении 97,52,5 получают 77 мг (80 ) целевого продукта. Тонкослойная хроматография смесь дихлорметана и метанола в соотношении 91,0,42. Пример 1.2 п-аминофенил-3 метилфукозид 6 г (21 ммоль) п-нитрофенилфукозида в 300 мл абсол. метанола смешивают с 7,84 г (31,5 ммоль) окиси дибутилолова и нагревают с обратным холодильником в течение двух часов. Затем сгущают, остаток сушат и подают в 300 мл диметилформамида. После добавки 15,7 мл метилйодида реакционную смесь перемешивают в течение 40 часов при 70 С. Растворитель удаляют в вакууме и остаток подают в 300 мл дихлорметана. Суспензию фильтруют, остающийся раствор снова сгущают и подвергают флеш-хроматографии 18 5307 1 с применением в качестве элюента смеси дихлорметана и метанола в соотношении 991. После сгущения получают 3815 мг (61 ) целевого продукта. 1.2) п-аминофенил-3 метилфукозид 3,81 г (12,73 ммоль) п-нитрофенил-3 метилфукозида гидрируют аналогично примеру 1.1. Выход 3 г (88 ). Тонкослойная хроматография смесь дихлорметана и метанола в соотношении 910,53. Пример 1.3 п-аминофенил-3-О-метилфукозид Соединение получают аналогично примеру 1.2 из п-нитрофенилфукозида. Выход 63 через две стадии. Тонкослойная хроматография смесь дихлорметана и метанола в соотношении 910,39. Пример 1.4 п-аминофенил-4-О-метилфукозид 1 г (3,5 ммоль) п-нитрофенилфукозида в 100 мл абсол. тетрагидрофурана смешивают с 31 мг п-толуолсульфокислоты и 1134 мг (7 ммоль) триэтилортоацетата. После перемешивания в течение 15 минут при 20 С растворитель отгоняют в вакууме. Остаток подают в 50 мл тетрагидрофурана и 3 мл диметилформамида, после чего смешивают с 4165 мкл бензилбромида и 210 мг 60 -ного гидрида натрия. После перемешивания при 20 С в течение часа к смеси добавляют 10 мл 80 -ной уксусной кислоты, сгущают и остаток очищают путем флеш-хроматографии с применением в качестве элюента смеси дихлорметана и метанола в соотношении 991. После сгущения и сушки получают 1236 мг 1000 мг (2,39 ммоль) п-нитрофенил-2 бензил-4 ацетилфукозида растворяют в 60 мл бензола. После добавки 2988 мкл метилйодида и 1109 мг окиси серебра реакционную смесь нагревают с обратным холодильником в течение 8 часов. Полученную смесь разделяют на компоненты путем флеш-хроматографии с применением в качестве элюента смеси дихлорметана и метанола в соотношении 991. Выделяют 239 мг (23 ) пнитрофенил-2-О-бензил-3-О-ацетил-4 метилфукозида и 653 мг (63 ) изомерного п-нитрофенил-2-О-бензил-3-О-метил-4 ацетилфукозида в виде белого твердого вещества. 1.4) п-аминофенил-4 метилфукозид 224 мг (0,52 ммоль) п-нитрофенил-2 бензил-3 ацетил-4-О-метилфукозида растворяют в 20 мл метанола и смешивают с 390 мкл 1-н. раствора метилата натрия. После перемешивания в течение 16 часов при 20 С нейтрализуют 80 -ной уксусной кислотой, сгущают и подают в дихлорметан. Органическую фазу промывают 1-н. раствора бикарбоната натрия и водой, сушат и сгущают. Остаток подают в 20 мл метанола и гидри 19 5307 1 руют над палладием на активном угле аналогично примеру 1.1. После сгущения продукт подают в воду и подвергают лиофилизации. Выделяют 119 мг (88 ) белого аморфного твердого вещества. Тонкослойная хроматография смесь дихлорметана и метанола в соотношении 910,38. Пример 1.5 п-аминофенил-3-О-н-пропилфукозид Аналогично примеру 1.4.б) соединение примера 1.4.а) подвергают взаимодействию с пропилйодидом с получением изомерных продуктов 3- и 4-пропилирования. В результате разделения путем хроматографии получают п-нитрофенил-2-О-бензил-3 н-пропил-4-ацетилфукозид (выход 49 ) и п-нитрофенил-2 бензил-3 ацетил-4 н-пропилфукозид (выход 29 ). 1.5.б) п-аминофенил-3 н-пропилфукозид Фракцию 1 примера 1.5.а) подвергают реакции аналогично примеру 1.4 с получением 78 целевого соединения. Тонкослойная хроматография смесь дихлорметана и метанола в соотношении 910,42. Пример 1.6. п-аминофенил-3-десоксифукозид 1 г (3,5 ммоль) п-нитрофенилфукозида в 100 мл тетрагидрофурана смешивают с 31 мг п-толуолсульфокислоты и 1134 мг (7 ммоль) триэтилортоацетата. После перемешивания в течение 15 минут при 20 С растворитель отгоняют в вакууме. Добавляют 100 мл насыщенного раствора хлористого водорода в дихлорметане. После 10-минутной реакции сгущают и продукт очищают путем флеш-хроматографии с применением в качестве элюента смеси дихлорметана и метанола в соотношении 991. Получают 793 мг (65 ) целевого продукта. Тонкослойная хроматография смесь дихлорметана и метанола в соотношении 97,52,50,36. 1.6.б) п-нитрофенил-3,6-дидесокси-3-хлоргулозид 375 мг (1,08 ммоль) п-нитрофенил-3,6-дидесокси-3-хлор-4 ацетилгулозида растворяют в 25 мл метанола и смешивают с 10 каплями 1-н. раствора метилата натрия. По истечении 20 минут подкисляют уксусной кислотой, сгущают и распределяют между 400 мл дихлорметана и 60 мл воды. Органическую фазу сушат, сгущают и осаждают из смеси дихлорметана и диэтилового эфира. Получают 315 мг (96 ) целевого продукта. 1.6.) п-аминофенил-3-десоксифукозид 315 мг (1,04 ммоль) п-нитрофенил-3,6-дидесокси-3-хлоргулозида растворяют в 40 мл метанола, смешивают с 200 мг палладия на активном угле и 290 мкл триэтиламина,20 5307 1 после чего смесь гидрируют в течение 4 дней в атмосфере водорода при незначительном избыточном давлении. Суспензию фильтруют, промывают, сгущают и продукт очищают путем флеш-хроматографии с применением в качестве элюента смеси дихлорметана и метанола в соотношении 97,52,5. Получают 160 мг (65 ) десокси-соединения. Тонкослойная хроматография смесь дихлорметана и метанола в соотношении 9550,18. Пример 1.7 п-аминофенил-3,4-дидесоксифукозид 400 мг (1,16 ммоль) п-нитрофенил-3,6-дидесокси-3-хлор-4 ацетилгулозида примера 1.6.а) растворяют в 55 мл метанола, смешивают с 323 мкл триэтиламина и гидрируют в атмосфере водорода при незначительном избыточном давлении над 10 -ным палладием на активном угле. После перемешивания в течение 16 часов при 20 С фильтруют на силикате марки , промывают, сгущают и подают в 100 мл метанола. К смеси добавляют 1,5 мл 1-н. раствора метилата натрия и перемешивают при комнатной температуре в течение 16 часов. Нейтрализуют уксусной кислотой, сгущают и полученные продукты разделяют путем флеш-хроматографии с применением в качестве элюента смеси дихлорметана и метанола в соотношении 97,52,5. После сгущения соответствующих фракций и переосаждения из смеси метанола и диэтилового эфира получают 120 мг (46 ) целевого соединения. Тонкослойная хроматография смесь дихлорметана и метанола в соотношении 9550,31 и 77 мг (28 ) п-аминофенил-3-десоксифукозида. Тонкослойная хроматография смесь дихлорметана и метанола в соотношении 9550,18. Пример 1.8 п-аминофенил-3,4-эпоксифукозид 80 мг (0,23 ммоль) п-нитрофенил-3,6-дидесокси-3-хлор-4-О-ацетилгулозида примера 1.6.а) подают в 10 мл метанола и смешивают с 345 мкл 1-н. раствора метилата натрия. После ультразвуковой обработки в течение часа подкисляют 80 -ной уксусной кислотой, сгущают и подвергают хроматографии с применением в качестве элюента смеси дихлорметана и метанола в соотношении 991. После сгущения соответствующих фракций продукт подают в метанол и гидрируют над палладием на активном угле аналогично примеру 1.1. Получают 46 мг (75 ) целевого соединения. Масс-спектр (бомбардировка быстрыми атомами, далее бба) /е 238 М 1. Пример 1.9 п-аминофенил-4-десоксифукозид Данное соединение получают аналогично методу авторов Т.и .в источнике . . 209 (1991 г.), стр. 119, исходя из п-нитрофенилфукозида через п-нитрофенил-2,3-дибензоил-4,6-дидесокси-4-йодфукозид. Тонкослойная хроматография смесь дихлорметана и метанола в соотношении 90100,3. 21 1 г (3,5 ммоль) п-нитрофенилфукозида и 1,3 г (5,2 ммоль) окиси дибутилолова нагревают с обратным холодильником в 50 мл метанола в течение 2 часов. Раствор сгущают, остаток подают в 50 мл диоксана, смешивают с 2 мл сложного метилового эфира бромуксусной кислоты и 100 мг йодида тетрабутиламмония и нагревают с обратным холодильником в течение 16 часов. Растворитель упаривают и продукт очищают путем флеш-хроматографии с применением в качестве элюента смеси дихлорметана и метанола в соотношении 991. После сгущения соответствующих фракций и переосаждения из смеси метанола и диэтилового эфира получают 455 мг (37 ) целевого соединения. 1.10) п-аминофенил-3 карбоксиметилфукозид 282 мг (0,79 ммоль) п-нитрофенил-3-метоксикарбонилметилфукозида растворяют в 20 мл метанола и смешивают с 440 мкл 2-н. раствора гидроокиси лития. После перемешивания при 20 С в течение 2 часов доводят до значения рН 3 с помощью кислого ионообменника марки 108 устанавливают и фильтруют. К фильтрату добавляют 250 мг палладия на активном угле. Затем гидрируют в атмосфере водорода при незначительном избыточном давлении в течение 1,5 часов, катализатор отделяют и промывают метанолом. Продукт сгущают, подают в воду и подвергают сублимационной сушке. Получают 212 мг(86 ) целевого продукта. Тонкослойная хроматография смесь ацетонитрила, воды и ледяной уксусной кислоты в соотношении 510,20,24. Пример 1.11 п-аминофенил-3 метоксикарбонилметилфукозид 250 мг (0,7 ммоль) п-нитрофенил-3-метоксикарбонилметилфукозида примера 1.10.а) растворяют в 20 мл метанола и гидрируют в атмосфере водорода при незначительном избыточном давлении в течение 1,5 часов. Катализатор отделяют и промывают метанолом. Продукт сгущают, подают в воду и подвергают сублимационной сушке. Получают 195 мг (85 ) целевого продукта. Тонкослойная хроматография смесь дихлорметана и метанола в соотношении 910,43 масс-спектр (бба) /е 328 М 1. Пример 1.12 п-аминофенил-3 гидроксиэтилфукозид 1000 мг (2,8 ммоль) п-нитрофенил-3-метоксикарбонилметилфукозида растворяют в смеси 160 мл тетрагидрофурана и 40 мл воды и смешивают с 53 мг борана натрия. По истечении 10 минут растворитель упаривают и остаток очищают путем флешхроматографии с применением в качестве элюента смеси дихлорметана и метанола в соотношении 955. После сгущения соответствующих фракций, подачи в воду и сублимационной сушки получают 362 мг (40 ) целевого продукта. 1.12) п-аминофенил-3 гидроксиэтилфукозид После гидрирования 362 мг соединения примера 1.12.а) аналогично примеру 1.1. получают 270 мг (82 ) целевого продукта. Тонкослойная хроматография смесь ацетонитрила и воды в соотношении 1010,43. Пример 1.13 п-аминофенил-2 карбоксиметилфукозид 250 мг (0,88 ммоль) п-нитрофенилфукозида растворяют в 25 мл абс. тетрагидрофурана и 3 мл диметилформамида. К смеси добавляют 80 мг (2,64 ммоль) 80 -ного гидрида натрия и, после 10-минутного перемешивания при 20 С, 35 мкл сложного бензилового эфира бромуксусной кислоты. Через каждые 10 минут 3 раза добавляют еще по 35 мкл сложного бензилового эфира бромуксусной кислоты. Перемешивают в течение 30 минут и обрабатывают метанолом. По истечении 10 минут подкисляют 5 мл 80 -ной уксусной кислоты. Сгущают и дополнительно перегоняют в присутствии дихлорметана. Очистку путем флеш-хроматографии начинают с применением в качестве элюента смеси дихлорметана, метанола и ледяной уксусной кислоты в соотношении 90101. Позже применяют ту же самую смесь, но в соотношении 80202. После сгущения соответствующих фракций остатки дигерируют диэтиловым эфиром и из первого элюента получают 157 мг(42 ) целевого соединения. Тонкослойная хроматография смесь ацетонитрила, воды и ледяной уксусной кислоты в соотношении 510,20,65. Из второго элюента получают 33 изомерного 3 алкилированного соединения. Тонкослойная хроматография смесь ацетонитрила, воды и ледяной уксусной кислоты в соотношении 510,20,54. 1.13) п-аминофенил-2 карбоксиметилфукозид Омыление и гидрирование 150 мг п-нитрофенил-2-О-метоксикарбоилметилфукозида осуществляют аналогично примеру 1.10. Получают 109 мг (83 ) целевого продукта. Тонкослойная хроматография смесь ацетонитрила, воды и ледяной уксусной кислоты в соотношении 510,20,35. Пример 1.14.а) Синтез региоизомерных продуктов моносукцинилирования п-нитрофенилфукозида 1100 мг (3,86 ммоль) п-нитрофенилфукозида растворяют в 50 мл пиридина и смешивают с 580 мг (5,79 ммоль) ангидрида янтарной кислоты. После перемешивания в течение 16 часов при 20 С сгущают и дважды дополнительно перегоняют в присутствии дихлорметана. Осаждают из смеси дихлорметана и диэтилового эфира и получают 1 г неразделимой смеси, которую подают в смесь метанола и воды и смешивают с 846 мг 5307 1 сутствии диметилформамида. Остаток подают в диметилформамид и смешивают с 618 мкл бензилбромида. После одночасовой ультразвуковой обработки бромид цезия отфильтровывают и фильтрат сгущают. Распределяют между 500 мл этилацетата и 50 мл воды. Органическую фазу сушат и сгущают. Компоненты разделяют путем флеш-хроматографии с применением в качестве элюента смеси дихлорметана и метанола в соотношении 991. Получают Фр. 1 87 мг (4,8 ) п-нитрофенил-3(3-бензилоксикарбонилпропионил)фукозида. Тонкослойная хроматография смесь дихлорметана и метанола в соотношении 9550,45. Фр. 2 27 мг (1,5 ) п-нитрофенил-2(3-бензилоксикарбонилпропионил)фукозида. Тонкослойная хроматография смесь дихлорметана и метанола в соотношении 9550,34. Фр. 3 190 мг (10,3 ) п-нитрофенил-4(3-бензилоксикарбонилпропионил)фукозида,Тонкослойная хроматография смесь дихлорметана и метанола в соотношении 9550,28. 1.14) п-аминофенил-3 сукцинилфукозид 85 мг (0,17 ммоль) фракции 1 примера 1.14.а) растворяют в 5 мл тетрагидрофурана и 1 мл воды. Добавляют 20 мг двуокиси платины и гидрируют в течение 8 часов. Катализатор отфильтровывают, промывают смесью тетрагидрофурана и воды и фильтрат сгущают. Остаток подают в воду и лиофилизуют. Получают 57 мг (94 ) целевого продукта. Тонкослойная хроматография смесь ацетонитрила, воды и ледяной уксусной кислоты в соотношении 510,20,65. Пример 1.15 п-аминофенил-2 сукцинилфукозид Фракцию 2 примера 1.14.а) гидрируют аналогично примеру 1.14. Выход 16 мг (87 ). Тонкослойная хроматография смесь ацетонитрила, воды и ледяной уксусной кислоты в соотношении 510,20,62. Пример 1.16 п-аминофенил-4 сукцинилфукозид 5307 1 Фракцию 3 примера 1.14.а) гидрируют аналогично примеру 1.14. Выход 125 мг(88 ). Тонкослойная хроматография смесь ацетонитрила, воды и ледяной уксусной кислоты в соотношении 510,20,63. Пример 1.17 п-аминофенил-3,4-диметилфукозид 377 мг (1,16 ммоль) соединения примера 1.1.а) растворяют в 30 мл абс. тетрагидрофурана и смешивают последовательно с 690 мкл бензилбромида и 52 мг гидрида натрия, после чего перемешивают при 20 С. Через 4 часа и 6 часов еще раз добавляют 690 мкл бензилбромида и гидрида натрия. Реакционную смесь перерабатывают аналогично примеру 1.1.б). Получают 245 мг (51 ) целевого соединения. 1.17.б) п-нитрофенил-2 бензил-3,4-диметилфукозид 245 мг (0,59 ммоль) п-нитрофенил-2-О-бензил-3,4 изопропилиденфукозида перемешивают в 80 -ной уксусной кислоте в течение 16 часов при 20 С. Сгущают и остаток перемешивают со смесью диэтилового эфира и пентана. Отсасывают, сушат и полученный продукт подают в 20 мл абс. тетрагидрофурана. К смеси добавляют 45 мг 80 ного гидрида натрия и через 15 минут - 160 мкл метилйодида. Перемешивают в течение 20 часов при 20 С, после чего обрабатывают метанолом и ледяной уксусной кислотой,сгущают и остаток распределяют между дихлорметаном и водой. Органическую фазу сушат, сгущают и очищают путем флеш-хроматографии с применением в качестве элюента смеси дихлорметана и метанола в соотношении 1001. Соответствующие фракции сгущают и сушат, после чего получают 188 мг (79 ) целевого продукта. 1.17) п-аминофенил-3,4-диметилфукозид 180 мг (0,45 ммоль) соединения примера 1.17.б) в смеси 15 мл метанола и 3 мл дихлорметана после добавления 50 мг палладия на активном угле гидрируют при комнатной температуре в течение 2 суток. Катализатор отфильтровывают, фильтрат сгущают и очищают путем флеш-хроматографии с применением в качестве элюента смеси дихлорметана и метанола в соотношении 97,52,5. Получают 86 мг (68 ) целевого соединения. Тонкослойная хроматография смесь дихлорметана и метанола в соотношении 9550,21. Пример 1.18 п-аминофенил-3 карбамоилметилфукозид 5307 1 воду и лиофилизуют. Получают 95 мг (колич.) целевого соединения. Тонкослойная хроматография смесь ацетонитрила и воды в соотношении 1010,43. Пример 1.19 п-аминофенил-2 гидроксиэтилфукозид 200 мг (0,56 ммоль) п-нитрофенил-2 метоксикарбонилметилфукозида примера 1.13.а) подвергают реакции аналогично примерам 1.12.а) и 1.12. Выход 76 мг (45 через 2 стадии). Тонкослойная хроматография смесь дихлорметана и метанола в соотношении 910,2. Пример 1.20 п-аминофенил-3,6-дидесокси-3-хлоргулозид 50 мг (0,165 ммоль) соединения примера 1.6.б) гидрируют в 5 мл метанола над палладием на активном угле в течение часа. Отфильтровывают от катализатора, промывают,сгущают, подают в воду и лиофилизуют. Получают 45 мг (89 ) целевого соединения. Тонкослойная хроматография смесь дихлорметана и метанола в соотношении 910,35. Пример 1.21 п-аминофенилрамнозид 300 мг п-нитрофенилрамнозида (сигма) подвергают реакции аналогично примеру 1.1 и получают 96 целевого соединения. Пример 1.22 п-аминофенил-3 карбоксиметилрамнозид 5307 1 течение 2 часов, сгущают и подают в 30 мл диоксана. К смеси добавляют 85 мг йодида тетрабутиламмония и 950 мкл сложного метилового эфира бромуксусной кислоты, после чего нагревают с обратным холодильником в течение 16 часов. В случае необходимости добавляют еще 1 мл сложного метилового эфира бромуксусной кислоты и продлевают время реакции. Сгущают и остаток очищают путем флеш-хроматографии. п-нитрофенил 3-О-метокси-карбонилметилрамнозид элюируют с применением смеси дихлорметана и метанола в соотношении 991. После сушки получают 408 мг (70 ) продукта. Тонкослойная хроматография смесь дихлорметана и метанола в соотношении 9550,36. Пример 1.22 п-аминофенил-3 карбоксиметилрамнозид п-нитрофенил-3 метоксикарбонилметилрамнозид подвергают реакции аналогично примеру 1.10 с получением 80 целевого продукта. Тонкослойная хроматография смесь ацетонитрила, воды и ледяной уксусной кислоты в соотношении 510,20,26. Пример 1.23 п-аминофенилгалактопиранозид 3,0 г (10 ммоль) п-нитрофенилгалактопиранозида растворяют в 50 мл смеси метанола и воды в соотношении 11. После добавления 200 мг 10 -ного палладия на активном угле гидрируют в атмосфере водорода при незначительном избыточном давлении. Суспензию фильтруют на силикате марки , после чего фильтруемый материал промывают 100 мл горячей смеси метанола и воды в соотношении 11. Фильтрат сгущают в вакууме. В результате перекристаллизации из метанола получают 2,11 г (78 ) бесцветных кристаллов. Тонкослойная хроматография метанол 0,62 20-39,5 (с 1,0/Н 2) т.п.166 С. Пример 1.24. п-аминофенил-2-О-метилгалактопиранозид Раствор 9,0 г (30 ммоль) п-нитрофенилгалактопиранозида, 16,7 г (60 ммоль) хлортрифенилметана и 609 мг (5 ммоль) ,-диметиламинопиридина в 100 мл абсолютного пиридина нагревают до 60 С в течение 4 часов. После сгущения в вакууме остаток очищают путем флеш-хроматографии с применением в качестве элюента смеси петролейного эфира и этилацетата в соотношении 2132, каждый раз в присутствии 0,5 триэтиламина. Получают 9,23 г (57 ) бесцветных кристаллов. Тонкослойная хроматография смесь дихлорметана, метанола и аммиака (25 ) в соотношении 1530,20,55 т.п.82 С. 8,7 г (16 ммоль) вышеуказанного соединения смешивают с 400 мл диметоксипропана и каталитическим количеством (400 мг, 1,7 ммоль) -камфора-10-сульфокислоты. После реакции при комнатной температуре в течение часа реакцию прекращают добавлением 240 мл (1,7 ммоль) триэтиламина и сгущают в вакууме. В результате флеш-хроматографии с применением в качестве элюента смеси петролейного эфира и этилацетата в соотношении 21 получают 6,2 г (66 ) бесцветной пены. Тонкослойная хроматография смесь петролейного эфира и этилацетата в соотношении 110,46 20-42,1(с 0,94/СН 2 С 2). 1.24.в) п-нитрофенил-2 метил-3,4 изопропилиден-6 трифенилметил-галактопиранозид 5,83 г (10 ммоль) соединения примера 1.24.б) растворяют в 100 мл диметилформамида и смешивают с 2,5 мл (40 ммоль) метилйодида, после чего к смеси порциями добавляют 450 мг (15 ммоль) 80 -ной суспензии гидрида натрия в минеральном масле. После реакции при комнатной температуре в течение 2 часов реакцию прекращают прикапыванием 10 мл метанола и сгущают в вакууме. Остаток подают в 1000 мл дихлорметана и раствор интенсивно перемешивают с 500 мл воды. Органическую фазу сушат над 50 г сульфата магния, сгущают в вакууме и очищают путем флеш-хроматографии с применением в качестве элюента смеси петролейного эфира и этилацетата в соотношении 12181. Получают 4,72 г (79 ) бесцветной пены. Тонкослойная хроматография смесь петролейного эфира и этилацетата в соотношении 110,72 20-35,7 (с 1,0/СН 3 ОН). 1.24.г) п-нитрофенил-2 метилгалактопиранозид Раствор 4,48 г (7,5 ммоль) вышеприведенного соединения в 200 мл дихлорметана смешивают с 20 мл 99 -ной трифторуксусной кислоты и перемешивают при комнатной температуре в течение 3 часов. После сгущения в вакууме остаток очищают путем флешхроматографии с применением в качестве элюента смеси петролейного эфира и этилацетата в соотношении 5121. Получают 1,09 г (46 ) бесцветных кристаллов. Тонкослойная хроматография смесь дихлорметана, метанола и аммиака (25 ) в соотношении 1530,20,42 т.п.177 С. 1.24) п-аминофенил-2 метилгалактопиранозид. 946 мг (3 ммоль) соединения примера 1.24.г) растворяют в 50 мл метанола и после добавления 0,5 мл воды и около 200 мг основного никеля Ренея гидрируют в атмосфере водорода при незначительном избыточном давлении в течение 2 часов. Суспензию фильтруют на силикате марки , после чего фильтруемый материал интенсивно промывают 100 мл метанола. После сгущения фильтрата в вакууме получают 579 мг (68 ) коричневатой пены. Тонкослойная хроматография смесь дихлорметана, метанола и аммиака (25 ) в соотношении 1530,20,28 20-39,3 (с 0,15/СН 3 ОН). Пример 1.25 п-аминофенил-3-О-метилгалактопиранозид Раствор 1,5 г (5,0 ммоль) п-нитрофенилгалактопиранозида в 40 мл абсолютного метанола смешивают с 1,87 г (7,5 ммоль) окиси дибутилолова и нагревают с обратным хо 28 5307 1 лодильником. Через 3 часа сгущают в вакууме и остаток сушат в течение часа в вакууме масляного насоса. Продукт подают в 40 мл абсолютного диоксана, полученный раствор смешивают с 1,9 мл (30 ммоль) метилйодида и реакционную смесь перемешивают в течение 16 часов при температуре ванны 100 С. Затем растворитель отгоняют в вакууме и остаток очищают путем флеш-хроматографии с применением в качестве элюента сначала смеси этилацетата и петролейного эфира в соотношении 21 а потом этилацетата. Получают 1,32 г (84 ) бесцветных кристаллов. Тонкослойная хроматография смесь дихлорметана, метанола и аммиака (25 ) в соотношении 1530,20,34 т.п.196 С 20-53,3 (с 1,0/СН 3 ОН). 1.25) п-аминофенил-3 метилгалактопиранозид 946 мг (3 ммоль) вышеприведенного соединения восстанавливают описанным в примере 1.24 образом и перерабатывают. Получают 656 мг (77 ) коричневатых кристаллов. Тонкослойная хроматография смесь дихлорметана, метанола и аммиака (25 ) в соотношении 1530,20,21 т.п.196 С 20-25,2 (с 1,0/СН 3 ОН). Пример 1.26 Ацетат п-аминофенил-4 метилгалактопиранозида Раствор 1,5 г (5,0 ммоль) п-нитрофенилгалактопиранозида в 40 мл абсолютного диоксана смешивают с 1,87 г (7,5 ммоль) окиси дибутилолова и нагревают с обратным холодильником. Через 3 часа полученный раствор смешивают с 3,6 мл (30 ммоль) бензилбромида и реакционную смесь перемешивают в течение 48 часов с обратным холодильником. Растворитель отгоняют в вакууме и остаток очищают путем флешхроматографии с применением в качестве элюента смеси этилацетата и петролейного эфира в соотношении 21 11. Получают 1,58 г (81 ) бесцветных кристаллов. Тонкослойная хроматография смесь дихлорметана, метанола и аммиака (25 ) в соотношении 1530,20,69 т.п.127 С. 1.26.б) п-нитрофенил-3 бензил-4,6 изопропилиденгалактопиранозид 6,26 г (16 ммоль) соединения примера 1.26.а) подвергают описанной в примере 1.24.б) реакции. После очистки путем флеш-хроматографии с применением в качестве элюента смеси петролейного эфира и этилацетата в соотношении 5131 получают 6,54 г (95 ) бесцветной пены. Тонкослойная хроматография смесь петролейного эфира и этилацетата в соотношении 110,34 20-38,9 (с 1,0/СН 2 С 2). 1.26.в) п-нитрофенил-2,3-дибензил-4,6 изопропилиденгалактопиранозид 4,31 г (10 ммоль) соединения примера 1.26.б) растворяют в 100 мл диметилформамида, после чего смешивают с 12 мл (100 ммоль) бензилбромида и порциями - 450 мг (15 ммоль) 80 -ной суспензии гидрида натрия в минеральном масле. После реакции при комнатной температуре в течение 2 часов реакцию прекращают прикапыванием 10 мл метанола и сгущают в вакууме. Остаток подают в 1000 мл дихлорметана и раствор интенсивно перемешивают с 500 мл воды. Органическую фазу сушат над 50 г сульфата магния,сгущают в вакууме и очищают путем флеш-хроматографии с применением в качестве элюента смеси петролейного эфира и этилацетата в соотношении 201151101. Получают 2,72 г (52 ) бесцветного масла, которое содержит еще примеси. Тонкослойная хроматография смесь петролейного эфира и этилацетата в соотношении 110,62. 29 2,6 г (5 ммоль) вышеуказанного соединения подвергают описанной в примере 1.24.г) реакции. После сгущения в вакууме и кипячения остатка в присутствии диэтилового эфира получают 805 мг (33 ) бесцветных кристаллов. Тонкослойная хроматография смесь петролейного эфира и этилацетата в соотношении 110,23 т.п.160 С. 1.26.д) п-нитрофенил-2,3-дибензил-6 трифенилметилгалактопиранозид 722 мг (1,5 ммоль) соединения примера 1.26.г) подвергают описанной в примере 1.24.а) реакции. После очистки путем флеш-хроматографии с применением в качестве элюента смеси петролейного эфира и этилацетата в соотношении 15110151 получают 880 мг (81 ) бесцветной пены. Тонкослойная хроматография смесь петролейного эфира и этилацетата в соотношении 210,79 20-25,3 (с 0,3/СН 2 С 2). 1.26.е) п-нитрофенил-2,3-дибензил-4 метил-6 трифенилметилгалакто пиранозид 724 мг (1 ммоль) вышеуказанного соединения подвергают описанному в примере 1.24.в) метилированию. После очистки путем флеш-хроматографии с применением в качестве элюента смеси петролейного эфира и этилацетата в соотношении 10151 получают 662 мг (90 ) бесцветной пены. Тонкослойная хроматография смесь петролейного эфира и этилацетата в соотношении 510,66 20-38,7 (с 0,2/22). 1.26) Ацетат п-аминофенил-4 метил-6 трифенилметилгалактопиранозида 590 мг (0,8 ммоль) вышеуказанного соединения растворяют в 50 мл 90 -ной уксусной кислоты. После добавки 200 мг 10 -ного палладия на активном угле гидрируют в течение 16 часов в атмосфере водорода при незначительном избыточном давлении. Суспензию фильтруют на силикате маркии фильтруемый материал интенсивно промывают 100 мл метанола. После сгущения фильтрата в вакууме и переосаждения остатка из смеси диэтилового эфира и петролейного эфира получают 253 мг (92 ) бесцветных кристаллов. Тонкослойная хроматография смесь дихлорметана и петролейного эфира в соотношении 510,12. Пример 1.27 п-аминофенил-6 метилгалактопиранозид 1.27.а) Бензилирование соединения примера 1.24.б) 5,84 г (10 ммоль) соединения примера 1.24.б) бензилируют описанным в примере 1.26.в) образом. После очистки путем флеш-хроматографии с применением в качестве элюента сначала смеси петролейного эфира и этилацетата в соотношении 15112151, а потом этилацетата, каждый раз в присутствии 0,5 триэтиламина, получают две фракции продукта Фракция 1 1,71 г (25 ) п-нитрофенил-2 бензил-3,4-О-изопропилиден-6-О-трифенилметилгалактопиранозида в виде желтоватой пены. Тонкослойная хроматография смесь петролейного эфира и этилацетата в соотношении 210,72 20-8,1(с 1,0/СН 2 С 2). Фракция 2 806 мг (19 ) п-нитрофенил-2-О-бензил-3,4-О-изопропилиденгалактопиранозида в виде желтоватого масла. Тонкослойная хроматография смесь петролейного эфира и этилацетата в соотношении 210,45 202,8(с 1,2/СН 3 ОН). 30
МПК / Метки
МПК: C07H 15/203, C07K 9/00, C07D 401/04, A61K 38/14, C07D 487/04, A61P 35/00
Метки: цитостатические, средства, модифицированные, углеводами
Код ссылки
<a href="https://by.patents.su/30-5307-modificirovannye-uglevodami-citostaticheskie-sredstva.html" rel="bookmark" title="База патентов Беларуси">Модифицированные углеводами цитостатические средства</a>
Способ получения производных гуанина или их кислотно-аддитивных фармацевтически приемлемых солей

Номер патента: 1330
Опубликовано: 16.09.1996
Авторы: Томас Энтони Креницки, Лилиа Мари Бьючэмп
МПК: A61K 31/52, C07D 473/18
Метки: гуанина, солей, приемлемых, производных, способ, фармацевтически, получения, или, кислотно-аддитивных
Текст:
...метанола и дид ХЛОРМЕТЗНЗ В РЗЗПИЧЪЕЫХ СООТНОШЕНИЯХ(0-15)ъ В результате получают целеВОЗ СОЕДИНЕНИЕвэтил 1 Кбензилоксн)карбонил-ВЪ-ва лината, 317 мг 5-ного палладиевого катализатора на угле, 100 мл метанола, 100 мл ТГФ и 18 мл 0,5 М соляной кислоты встряхивают в аппарате Парра при давлениводорода 50 фунтов над кв.дюйм в течение дня. Послеэтого реакционную смесь фильтруют через слой целита и концентрируют, получая твердое вещество, которое...
Способ получения производного аспартама, пригодного в качестве подслащивающего средства

Номер патента: 4855
Опубликовано: 30.12.2002
Авторы: Клод НОФР, Жан-Мари ТИНТИ
МПК: C07K 5/075, A23L 1/236
Метки: получения, производного, способ, пригодного, аспартама, средства, качестве, подслащивающего
Текст:
...кремния и оксиде алюминия (20, 877-9), никель Ренея (22, 167-8), рутениевая чернь (32, 671-2), рутений на угле (28,147-6), гидроксид палладия на угле (21,291-1), оксид палладия (20, 397-1), родиевая чернь (26, 734-1), родий на угле (33, 017-5) или родий на оксиде алюминия (83720), также позволяют получать соединение согласно изобретению. Однако эти катализаторы оказываются либо менее активными, требуя особенно более высоких давлений...
Способ получения средства защиты растений

Номер патента: 1071
Опубликовано: 14.03.1996
Авторы: Косоногова Людмила Владиславовна, Шуцкая Ольга Викторовна, Царева Зоя Ивановна, Талапин Виталий Иванович, Наумова Галина Васильевна
МПК: A01N 61/00
Метки: растений, способ, получения, защиты, средства
Текст:
...препарата с концентрацией органических веществ 9,6,что составляет 92,6 от органической массы угля или 74,8 от сухой навески бурого угля.Для приготовления средства защиты растений от болезней в смеситель загружают 1,9 кг полученного гуминового препарата и 0,8 кг комплексоната меди. Компоненты перемешивают. Получают 2,7 кг средства при следуюЩСМ СООТНОШЕНИЕ КОМПОНЕНТОВ, ГУМИНОВЫЙ препарат - 70, комплексонат - 30 мас..Полученную смесь...
Способ получения оптически активного (+)4-деметоксидауномицинона

Номер патента: 1803
Опубликовано: 30.12.1997
Авторы: Уолтер Кабри, Сильвия Де Бернардинис, Франко Франкаланчи, Серджио Пенко
МПК: C07C 50/36, C07C 50/38
Метки: оптически, 4-деметоксидауномицинона, способ, получения, активного
Текст:
...мл (755 ммоля) трифторметенсульфонильного анГИдриде. после чего реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Реакционную смесь затем охлаждали до 0 С и добавляли 5 л диметиленхлорида И 3 л 1 О-ной хлористоводородной кислоты. После отделения органическую фазу промывали водой. высушивали над сульфатом натрия и внпаривали растворительу ПРИ пониженном давлении, в результате че го было получено 13.75 г твердого вещества....
Способ получения -D-фенилтиоксилозидов

Номер патента: 478
Опубликовано: 30.03.1995
Авторы: Вероник Барберусс, Ежи Байгрович, Патрик Рено, Жан Мийе, Сот Самрет, Франсуа Беллами
МПК: A61K 31/70, C07H 13/04, C07H 15/203...
Метки: d-фенилтиоксилозидов, получения, способ
Текст:
...продукта. Т.пл. 175 С. .Согласно способу работы. описанному в получении 1. но исходя из 6.8 г (42.4-то моль) 2-нафталинтиола. 10.8 г (42.4103 моль) цианида ртути-(Н) Н 9(С)2 и 12 г(3 З.2Согласно способу работы, описанному в получении / но исходя из 5.8 г (13-109 моль)ПОСЛЕ перекристаллизации ИЗ СМЕСИ ЭТЗНО(пример 43). с Согласно способу работы, описанному в получении , но исходя из 5.58 г(32 г 1 О 3 моль 4-трифторметил-бензолтиола 8.87 г...
Предыдущий патент: Производные диоксида бензотиазина, фармацевтическая композиция, ингибирующая рецептор эндотелина, и способ ингибирования рецептора эндотелина
Следующий патент: Производные хиназолина, способы их получения и фармацевтическая композиция на их основе
Случайный патент: Способ получения трехкомпонентных комплексов малонилмочевины