Производные эритромицина, способ их получения и фармацевтическая композиция на их основе
Номер патента: 4361
Опубликовано: 30.03.2002
Авторы: Одиль ЛЕ МАРТРЕ, Алексис ДЕНИС, Янник БЕНЕДЕТТИ, Константин АГУРИДАС, Жан-Франсуа ШАНТО



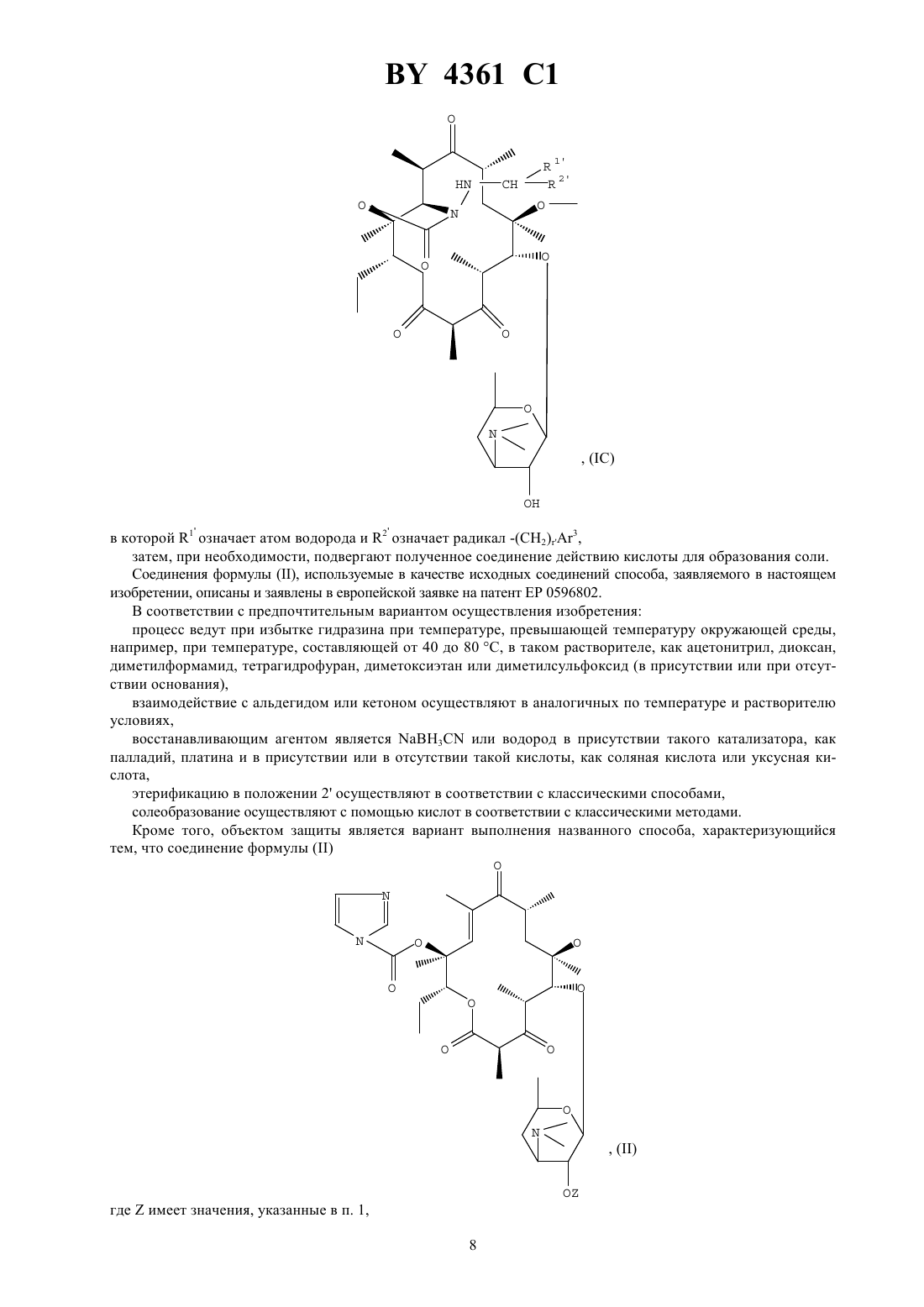
Текст
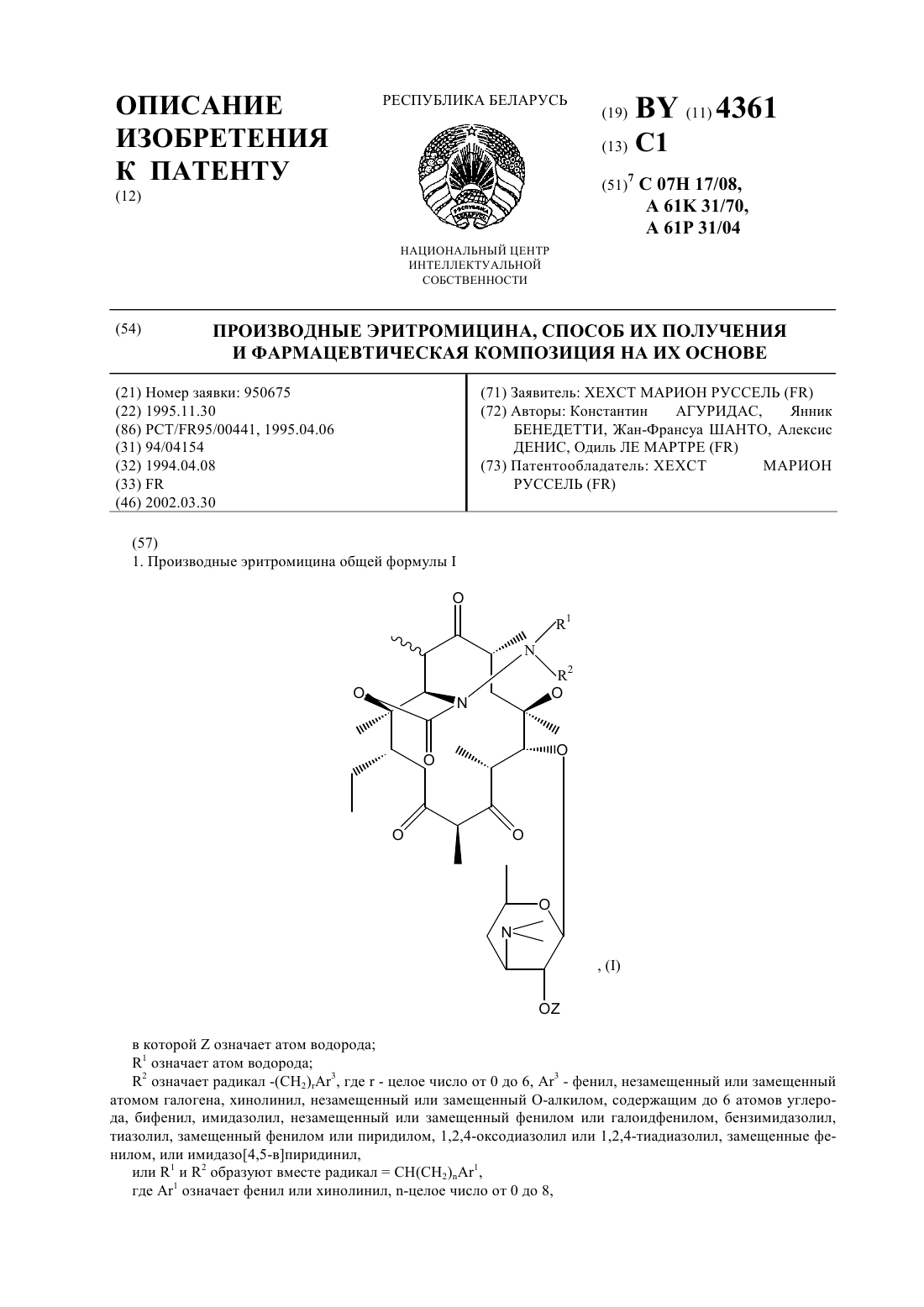
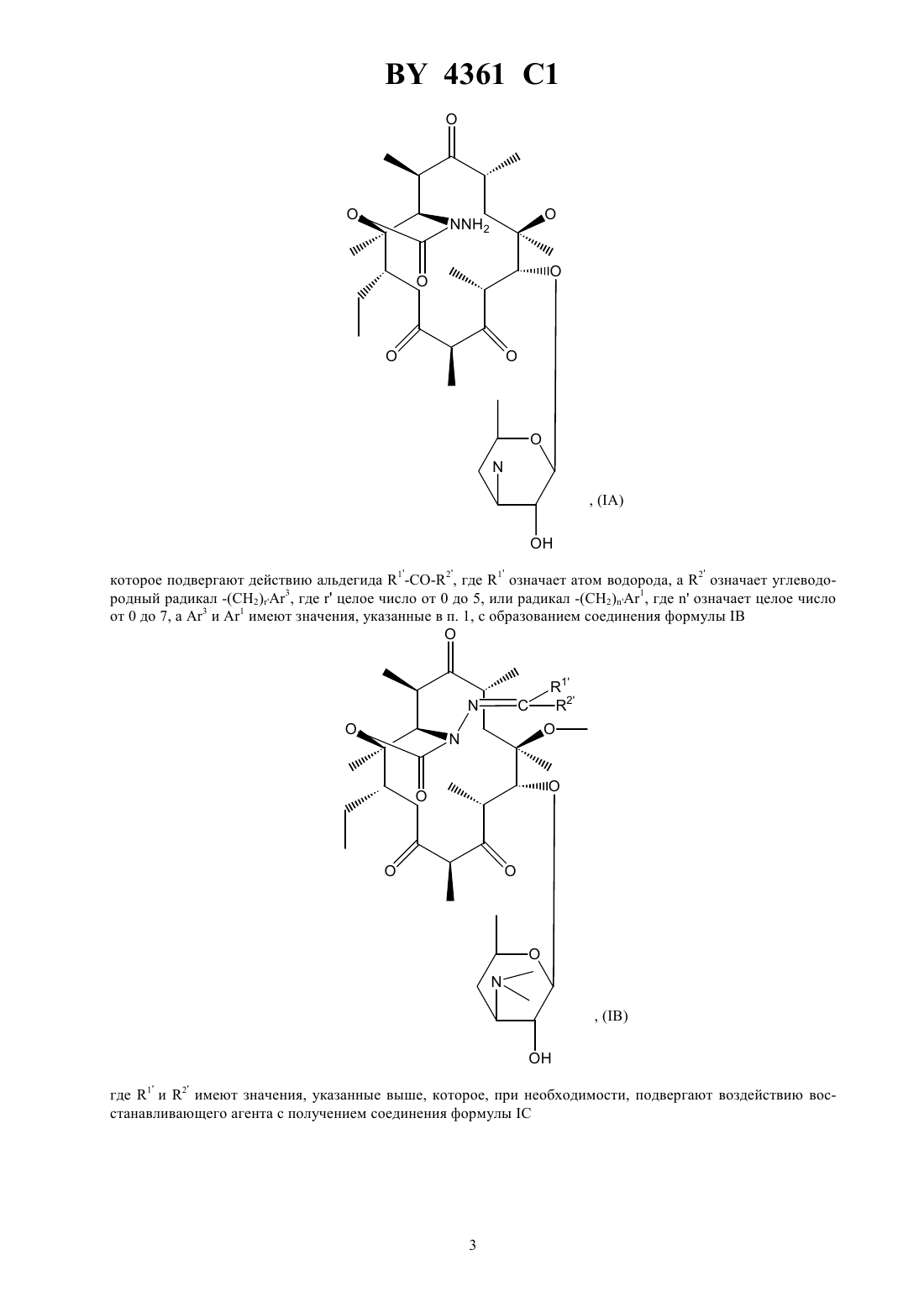
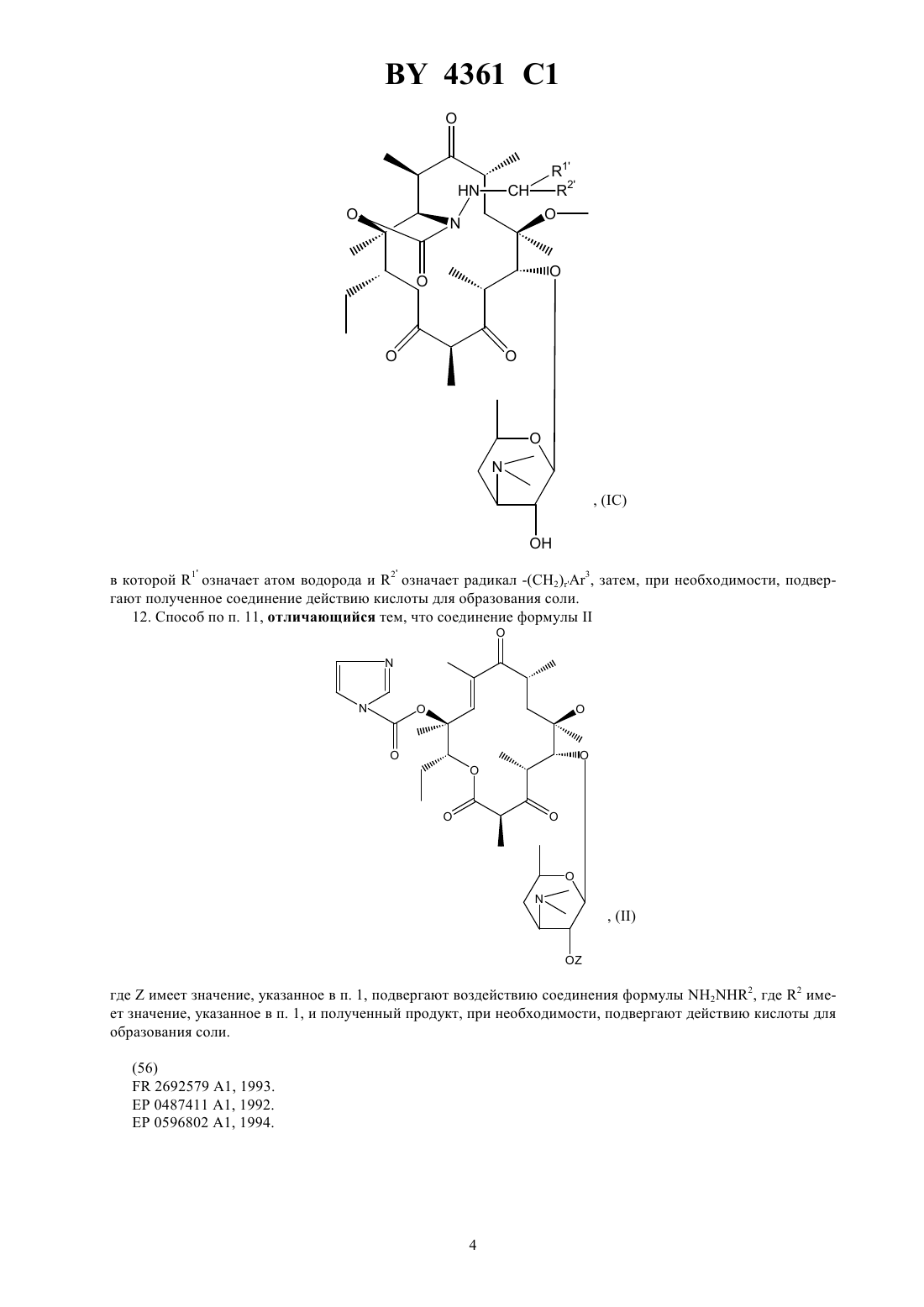
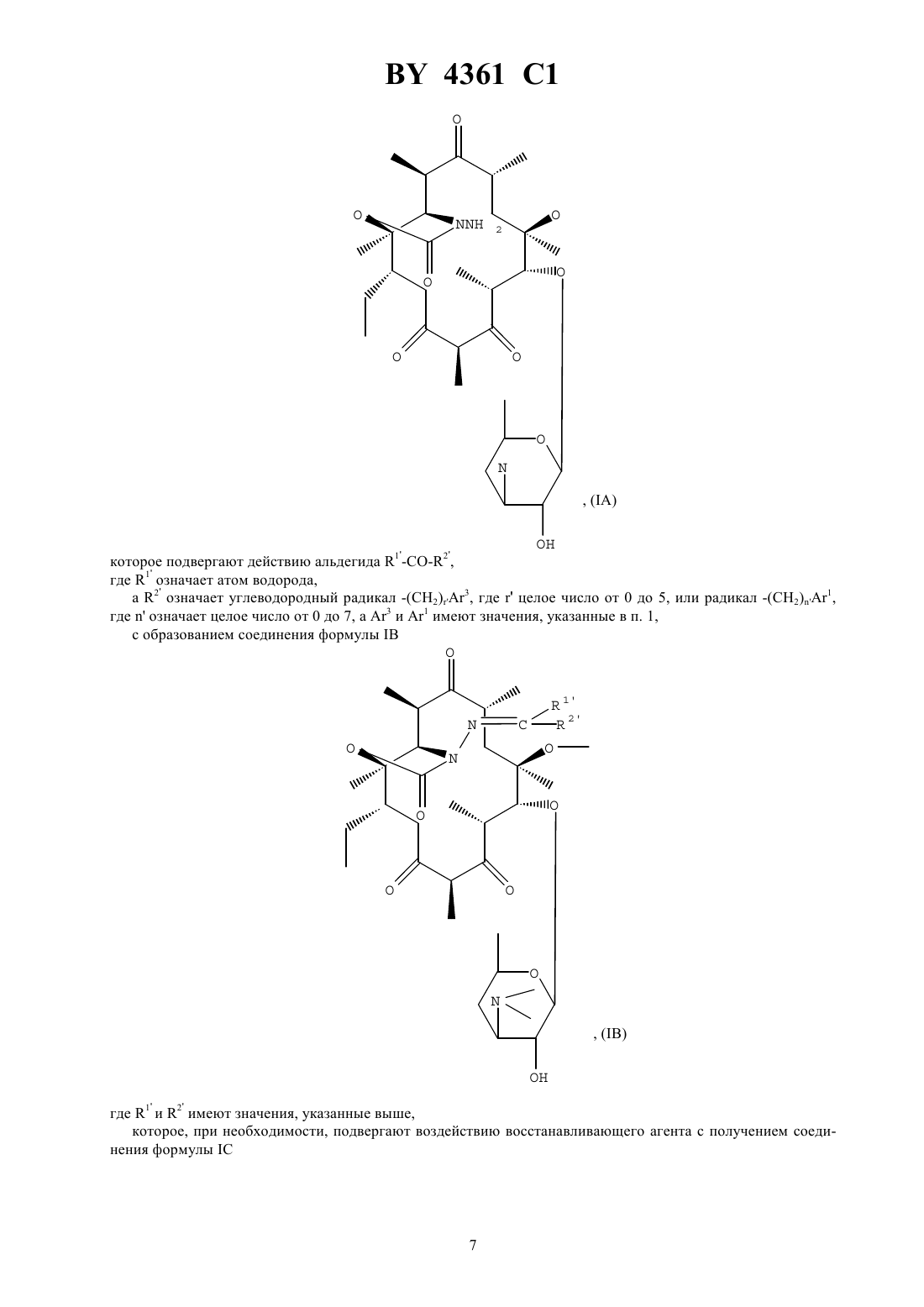
НАЦИОНАЛЬНЫЙ ЦЕНТР ИНТЕЛЛЕКТУАЛЬНОЙ СОБСТВЕННОСТИ ПРОИЗВОДНЫЕ ЭРИТРОМИЦИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ(71) Заявитель ХЕХСТ МАРИОН РУССЕЛЬ(73) Патентообладатель ХЕХСТ МАРИОН РУССЕЛЬ(57) 1. Производные эритромицина общей формулы в которойозначает атом водорода 1 означает атом водорода 2 означает радикал -(СН 2)3, где- целое число от 0 до 6, 3 - фенил, незамещенный или замещенный атомом галогена, хинолинил, незамещенный или замещенный -алкилом, содержащим до 6 атомов углерода, бифенил, имидазолил, незамещенный или замещенный фенилом или галоидфенилом, бензимидазолил,тиазолил, замещенный фенилом или пиридилом, 1,2,4-оксодиазолил или 1,2,4-тиадиазолил, замещенные фенилом, или имидазо 4,5-впиридинил,или 1 и 2 образуют вместе радикалСН(СН 2)1,где 1 означает фенил или хинолинил, -целое число от 0 до 8,4361 1 волнистая линия в положении 10 указывает, что метил может иметь конфигурациюили , или же представлен смесью конфигурациии ,или их кислотно-аддитивные соли. 2. Соединение формулыпо п. 1, в котором 3 представляет собой 4-хинолинил, возможно монозамещенный. 3. Соединение формулыпо п. 1, в котором 3 представляет собой незамещенный 4-хинолинил. 4. Соединение формулыпо п. 1, в котором 3 представляет собой замещенный группой метокси 4 хинолинил. 5. Соединение формулыпо п. 1, в котором 3 представляет собой тиазолил, замещенный пиридилом. 6. Соединение формулыпо любому из пп. 1 - 5, в которомпредставляет собой целое число от 1 до 4. 7. Соединение по п. 1, выбранное из группы, включающей 11,12-дидеокси-3-де 2,6-дидеокси-3-С-метил-3 метилрибогексопиранозил)окси)-6 метил-3 оксо-12,11-(оксикарбонил-(2-(3-(4-хинолинил)пропил)гидразоноэритромицин 11,12-дидеокси-3-де 2,6-дидеокси-3-С-метил-3 метилрибогексопиранозил)окси)-6 метил-3 оксо-12,11-(оксикарбонил-(2-(3-(7-метокси-4-хинолинил)пропил)гидразоноэритромицин 11,12-дидеокси-3-де 2,6-дидеокси-3-С-метил-3 метилрибогексопиранозил)окси)-6 метил-3 оксо-12,11-(оксикарбонил-(2-(3-(3-пиридинил-4-тиазолил)пропил)гидразоноэритромицин. 8. Соединения по любому из пп. 1 - 7 или их соли фармацевтически приемлемые кислотно-аддитивные соли, обладающие антибиотической активностью. 9. Соединения по п. 7 или их фармацевтически приемлемые кислотно-аддитивные соли, обладающие антибиотической активностью. 10. Фармацевтическая композиция, обладающая антибиотической активностью, содержащая действующее вещество и эксципиент, отличающаяся тем, что в качестве действующего вещества она содержит, по меньшей мере, одно соединение по п. 8 или 9. 11. Способ получения производных эритромицина общей формулыпо п. 1, характеризующийся тем, что соединение формулы гдеозначает атом водорода, подвергают воздействию гидразина 22 с получением соединения формулы которое подвергают действию альдегида 1 2 , где 1 означает атом водорода, а 2 означает углеводородный радикал -(СН 2)3, гдецелое число от 0 до 5, или радикал -(СН 2)1, гдеозначает целое число от 0 до 7, а 3 и 1 имеют значения, указанные в п. 1, с образованием соединения формулы где 1 и 2 имеют значения, указанные выше, которое, при необходимости, подвергают воздействию восстанавливающего агента с получением соединения формулы в которой 1 означает атом водорода и 2 означает радикал -(СН 2)3, затем, при необходимости, подвергают полученное соединение действию кислоты для образования соли. 12. Способ по п. 11, отличающийся тем, что соединение формулы гдеимеет значение, указанное в п. 1, подвергают воздействию соединения формулы 22, где 2 имеет значение, указанное в п. 1, и полученный продукт, при необходимости, подвергают действию кислоты для образования соли. 4361 1 Объектом защиты настоящего изобретения являются соединения общей формулы в которойозначает атом водорода 1 означает атом водорода 2 означает радикал -(СН 2)3, где- целое число от 0 до 6, 3 - фенил, незамещенный или замещенный атомом галогена, хинолинил, незамещенный или замещенный -алкилом, содержащим до 6 атомов углерода, бифенил, имидазолил, незамещенный или замещенный фенилом или галоидфенилом, бензимидазолил,тиазолил, замещенный фенилом или пиридилом, 1,2,4-оксодиазолил или 1,2,4-тиадиазолил, замещенные фенилом, или имидазо 4,5-впиридинил,или 1 и 2 образуют вместе радикалСН(СН 2)1,где 1 означает фенил или хинолинил, -целое число от 0 до 8,волнистая линия в положении 10 указывает, что метил может иметь конфигурациюили , или же представлен смесью конфигурациии ,или их кислотно-аддитивные соли. Среди предпочтительных соединений, согласно изобретению, можно назвать соединения формулы , в которой 3 представляет собой 4-хинолин, возможно монозамещенный по одному и/или другому из 2 хинолиновых циклов, например, те соединения, в которых 3 представляет собой незамещенный 4 хинолин, 4-хинолин, замещенный метокси группой, или тиазолил, замещенный пиридинилом. Еще более конкретно, объектом защиты являются соединения формулы , в которойпредставляет собой целое число от 1 до 4. Среди предпочтительных соединений, согласно заявляемому изобретению, можно назвать те соединения,получение которых приведено ниже в экспериментальной части описания, а более конкретно, соединения,выбранные из группы, включающей 11,12-дидеокси-3-де-2,6-дидеокси-3-С-метил-3-О-метилрибогексопиранозил)-окси)-6-О-метил-3 оксо-12,11-(оксикарбонил-(2-(3-(4-хинолинил)-пропил)-гидразоно-эритромицин,11,12-дидеокси-3-де-2,6-дидеокси-3-С-метил-3-О-метилрибогексопиринозил)-окси)-6-О-метил-3 оксо-12,11-(оксикарбонил-(2-(3-(7-метокси-4-хинолинил)-пропил)-гидразоно-эритромицин,11,12-дидеокси-3-де-2,6-дидеокси-3-С-метил-3-О-метилрибогексопиранозил)-окси)-6-О-метил-3 оксо-12,11-(оксикарбонил-(2-(3-(3-пиридинил-4-тиазолил)-пропил)-гидразоно-эритромицин. Соединения общей формулыобладают очень высокой антибиотической активностью на грамположительные бактерии, такие как стафилококки, пневмококки. Таким образом, соединения, согласно изобретению, могут быть использованы в качестве лекарственных средств при лечении микробных инфекций, а именно стафилококковых инфекций, например, стафилококковых септицемий, стафилококковых высыпаний на лице и коже, пиодермитов, септических или гнойных ран,фурункулов, карбункулов, флегмон, рожистых заболеваний и угрей таких стафилококковых заболеваний,как обычный острый тонзиллит или послегрипповая ангина, бронхопневмония, легочные нагноения таких стрептококковых заболеваний, как острые тонзиллиты, отиты, синуситы, скарлатина, таких пневмококковых заболеваний, как пневмонии, бронхиты бруцеллеза, дифтерии, гонококковых заболеваний. 4361 1 Соединения, заявляемые в настоящем изобретении, также активны по отношению к инфекциям, вызванным такими микробами, как, ,, , , ,или микробами рода . Кроме того, объектом защиты являются также в качестве лекарственных средств, а конкретнее лекарствантибиотиков, соединения формулы , указанные выше, а также их соли присоединения с минеральными или органическими фармацевтически пригодными кислотами. Более конкретно, настоящее изобретение относится в качестве лекарственных средств, а именно антибиотиков, к соединениям из примеров 5 или 11, 12 и 13 и их фармацевтически пригодным солям. Кроме того, объектом защиты настоящего изобретения являются фармацевтические композиции содержащие в качестве действующего вещества, по меньшей мере, одно лекарственное средство, указанное выше. Эти композиции могут быть введены оральным путем, ректально, парентерально или локально путем местных аппликаций на кожу и слизистую оболочку, однако, предпочтительным является оральное введение этих композиций. Композиции могут представлять собой жидкости или твердые вещества и иметь фармацевтические формы, обычно используемые при лечении человека, например простые или дражевидные таблетки, желатинозные капсулы, гранулы, свечи, препараты для инъекций, мази, кремы, гели эти композиции получены в соответствии с обычными методами получения. Действующее или действующие вещества могут находиться в этих фармацевтических композициях в смеси с обычно используемыми эксципиентами, например с тальком, гуммиарабиком, лактозой, крахмалом, стеаратом магния, масло-какао, водными или неводными лекарственными основами, животными или растительными жирами, парафиновыми производными,гликолями, различными смачивающими агентами, диспергаторами или эмульгаторами, консервантами. Эти композиции могут также иметь вид порошков, предназначенных для растворения перед использованием в соответствующем эксципиенте, например, в стерильной апирогенной воде. Вводимая доза изменяется в зависимости от заболевания, излечиваемого больного, способа введения лекарственного препарата и выбранного соединения. Доза может составлять, например, от 50 до 300 мг в день при введении орально взрослому человеку соединения из примера 5. Кроме того, объектом защиты настоящим изобретением является способ получения соединений формулы, характеризующийся тем, что соединение формулы гдеозначает атом водорода, подвергают воздействию или же гидразином 22 с получением соединения формулы (А) которое подвергают действию альдегида 1 2 , где 1 означает атом водорода, а 2 означает углеводородный радикал -(СН 2)3, гдецелое число от 0 до 5, или радикал -(СН 2)1,гдеозначает целое число от 0 до 7, а 3 и 1 имеют значения, указанные в п. 1,с образованием соединения формулы где 1 и 2 имеют значения, указанные выше,которое, при необходимости, подвергают воздействию восстанавливающего агента с получением соединения формулы в которой 1 означает атом водорода и 2 означает радикал -(СН 2)3,затем, при необходимости, подвергают полученное соединение действию кислоты для образования соли. Соединения формулы , используемые в качестве исходных соединений способа, заявляемого в настоящем изобретении, описаны и заявлены в европейской заявке на патент ЕР 0596802. В соответствии с предпочтительным вариантом осуществления изобретения процесс ведут при избытке гидразина при температуре, превышающей температуру окружающей среды,например, при температуре, составляющей от 40 до 80 С, в таком растворителе, как ацетонитрил, диоксан,диметилформамид, тетрагидрофуран, диметоксиэтан или диметилсульфоксид (в присутствии или при отсутствии основания),взаимодействие с альдегидом или кетоном осуществляют в аналогичных по температуре и растворителю условиях,восстанавливающим агентом является 3 или водород в присутствии такого катализатора, как палладий, платина и в присутствии или в отсутствии такой кислоты, как соляная кислота или уксусная кислота,этерификацию в положении 2 осуществляют в соответствии с классическими способами,солеобразование осуществляют с помощью кислот в соответствии с классическими методами. Кроме того, объектом защиты является вариант выполнения названного способа, характеризующийся тем, что соединение формулы 4361 1 подвергают воздействию соединением формулы 22 с получением соединения формулы (А) которое, если желают, обрабатывают агентом, способным заменить атом водорода группына радикал 1, указанный выше, за исключением значения водорода, с получением соединения формулы (В) которое, если желают, подвергают воздействию агентом, обеспечивающим этерификацию по группе ОН в положении 2, или воздействию кислотой с образованием соли, при этом предпочтительные условия по температуре и давлению аналогичны указанным выше. Нижеследующие примеры иллюстрируют настоящее изобретение. Пример 1. 11,12-дидеокси-3-де-2,6-дидеокси-3-С-метил-3-О-метилрибогексопиранозил)-окси)-6-О-метил-3-оксо 12,11-(оксикарбонил-(гидрозоно)-эритромицин, изомер 10 и изомер 10. В 5 мл цианистого метила и 0,5 мл воды суспендируют 353 мг 2-ацетат-11-деокси-10,11-дидегидро-3-де(2,6-дидеокси-3-С-метил-3-О-метилрибогексопиранозил)-окси)-12(1 Н-имидазол-1-ил)-карбонил)-6-Ометил-3-оксо-эритромицина, полученного по методике, указанной в заявке на Европейский патент ЕР 0596802, и 0,097 мл гидразингидрата. Реакционную массу нагревают при температуре 60 С в течение 3 ч. 9 4361 1 Затем реакционную смесь выливают в воду, экстрагируют этилацетатом, промывают, сушат. Осуществляют хроматографию реакционной смеси на двуокиси кремния, используя в качестве элюанта смесь изопропилового эфира, триэтиламина и метанола (90/10/10). Получают 101 мг целевого продукта (продукт ),0,45,и 106 мг соответствующего продукта 10 (продукт В). Продукт А Анализрасчетноеобнаружено соединение (А) соединение (В) 59,31 59,3 59,3 С Н 8,51 8,4 8,4 Пример 2. 11,12-дидеокси-3-де-(2,6-дидеокси-3-С-метил-3-О-метилрибогексопиранизил)-окси)-6-О-метил-3 оксо-12,11-(оксикарбонил-(2-(3-фенилпропилиден)-гидразоно-эритромицин. Растворяют в 2 мл тетрагидрофурана на молекулярном сите (4 А) 285 мг продукта А, полученного в примере 1, и 156 мг 3-фенил-пропиональдегида. Прибавляют 100 молекулярного сита (4 А) и нагревают при температуре 60 С в течение 24 ч. Фильтруют, концентрируют и очищают путем хроматографии, осуществляемой на двуокиси кремния, используя в качестве элюанта смесь этилацетата и триэтиламина (96-4). Собирают фракции 0,41 и получают 330 мг целевого продукта 0,3. Анализ С Н-2 . Пример 3. 11,12-дидеокси-3-де-2,6-дидеокси-3-С-метил-3-О-метилрибогексопиранозил)-окси)-6-О-метил-3 окси-12,11-(оксикарбонил-(2-(3-фенилпропил)-гидразоно-эритромицин. Прибавляют 23 мг цианоборгидрида натрия (3) в раствор, содержащий 1,5 мл метанола, 88 мг соединения, полученного в примере 2, и 50 мкл уксусной кислоты. Осуществляют концентрирование, обработку этилацетатом, прибавление воды, а затем с помощью 2-тво гидроокиси натрия обеспечивают рН 8. Затем проводят декантирование, промывание насыщенным раствором хлорида натрия и высушивание. Полученный продукт подвергают хроматографии на двуокиси кремния (элюант простой изопропиловый эфир-метанол-триэтиламин, 90-10-10). Собирают фракции 0,33. Полученную смесь обрабатывают смесью простой эфир-пентан и фильтруют. После выпаривания получают 70 мг целевого соединения. Пример 4. 11,12-дидеокси-3-де-2,6-дидеокси-3-С-метил-3-О-метилрибагексопиранозил)-окси)-6-О-метил-3 оксо-12,11-(оксикарбонил-(2-(3-(4-хинолинил)-2(Е)-(пропенилиден)-гидразоно-эритромицин. При температуре окружающей среды перемешивают в течение 5 ч 125 мг соединения А, полученного в примере 1, 73 мг 4-хинолинилпропеналя, получение которого описано ниже, и 40 мкл уксусной кислоты. При пониженном давлении удаляют метанол, после чего остаток обрабатывают смесью метиленхлорид-вода. С помощью концентрированного раствора гидрата окиси аммония устанавливают рН 9. Затем осуществляют декантирование, высушивание на сульфате магния, фильтрование и выпаривание насухо. Получают 211 мг продукта, который подвергают хроматографии на двуокиси кремния, используя в качестве элюанта смесь метиленхлорид-метанол (92-8). Продукт с 0,4 сгущают в смеси этилацетат-пентан (1-1). Затем полученный продукт отжимают и промывают в минимуме смеси этилацетат-пентан, высушивают в сушильном шкафу при пониженном давлении. Таким образом получают 109 мг целевого продукта. К примеру 4. Приготовление 4-хинолинпропеналя. Растворяют в 80 мл метиленхлорида 3,9 г 4-хинолинкарбокс-альдегида. Раствор охлаждают до температуры 10 С 5 С, после чего в течение 1 ч 30 мин прибавляют, поддерживая температуру на уровне 10 С,8,3 г 3-(трифенилфосфин)-пропеналя (С 6 Н 5)3 РС-СНО. Реакционную смесь оставляют до тех пор, пока ее температура не возвратится на значение 20 С, после чего продолжат перемешивание в течение 24 ч. Затем снова охлаждают до температуры 10 С, прибавляют 0,4 г (С 6 Н 5)3 РС-СНО. Перемешивают еще в течение 3 часов при температуре окружающей среды. Выпаривают метиленхлорид и получают продукт, который подвергают хроматогафии на двуокиси кремния, используя в качестве элюанта смесь этилацетат-циклогексан (4-6). Выделяют 2,12 г целевого продукта. Тпл 90 С. Пример 5. 11,12-дидеокси-3-де-2,6-дидеокси-3-С-метил-3-О-метилрибогексопиранозил)-окси)-6-О-метил-3 оксо-12,11-(оксикарбонил-(2-(3-(4-хинилинил)-пропил)-гидразоно-эритромицин. Растворяют в 10 мл этилацетата 0,38 г соединения, полученного в примере 4, и 38 мг оксида платины. Гидрируют при интенсивном перемешивании в течение 24 ч. Полученный продукт фильтруют, промывают в этилацетате и выпаривают при пониженном давлении. Получают 0,375 г соединения, которое обрабатывают 5 мл метанола, 175 мкл уксусной кислоты и 90 мг боргидрида натрия. Перемешивают в течение 3 ч при температуре окружающей среды. Отгоняют метанол, а остаток обрабатывают смесью метиленхлорид-вода. С помощью 28 -ного раствора гидрата окиси аммония устанавливают рН 8-9. Затем реакционную массу декантируют, промывают водой, высушивают, фильтруют и выпаривают досуха. Получают 0,37 г продукта,который подвергают хроматографии на двуокиси кремния, используя в качестве элюанта смесь этилацетаттриэтиламин (96-4). Получают 127 мг соединения (0,25), которое отжимают, промывают и сушат. Получают 90 мг целевого продукта, Тпл 189 С. ЯМР 3 ррм, 300 Мгц 1,34- 1,486 и 12 СН 3 2,30(СН 3)2 2,656-ОСН 3 3,06 Н 4 3,1910 3,74 Н 11 5,50 ( переменная) -2 7,30 Н 3 хинолин 7,53-7,68 Н 6-Н 7 хинолин 8,10 Н 5-Н 8 хинолин 8,79 Н 2 хинолин. Пример 6. 11,12-дидеокси-3-де-2,6-дидеокси-3-С-метил-3-О-метилрибогексопиранозил)-окси)-6-О-метил-3-оксо 12,11-(оксикарбонил-(2-(3-(1 Н-бензимидазол-1-ил)-пропил)-гидразоно-эритромицин. Осуществляют перемешивание в течение 18 ч при температуре окружающей среды раствора 300 мг продукта, полученного в примере 1, 168 мг 3-имидозолилпропаноля (получение которого описано ниже) и 90 мл уксусной кислоты в 9 мл метанола, по истечении этого времени в реакционную смесь добавляют 40 мг цианоборгидрида натрия и осуществляют перемешивание еще в течение 5 ч, а затем добавляют 120 мг цианоборгидрида натрия и 200 мкл уксусной кислоты. Перемешивание продолжают в течение 48 ч, затем добав 11 4361 1 ляют смесь метиленхлорида и воды, с помощью 32 -ного раствора гидрата окиси аммония устанавливают рН 8-9, после чего отделяют органическую фазу, сушат и выпаривают насухо. Получают 0,6 г остатка, который подвергают хроматографии на двуокиси кремния (элюантом служит этилацетат-метанол-триэтиламин 92-6-2) полученный продукт сгущают в смеси простой эфир-пентан 1-5. Получают 143 мг целевого неочищенного продукта, который растворяют в 1 мл этилацетата, фильтруют и кристаллизуют путем прибавления 3 мл пентана. После высушивания получают 85 мг целевого продукта (Тпл 197 С). Анализ С 41 Н 635 О 10 785,98 С Н рассчитанный 62,65 8,08 8,91 обнаруженный 62,5 8,1 8,8. ЯМР 3 ррм 3,18 10 3,6911 0,8415 СН 3 3,862 2,45 -(СН 3)2 2,606 ОСН 3 5,562,652,81 -2- 4,50 -2- 7,26-8,02 5 бензимидазол. К примеру 6 Приготовление 3-имидазолилпропаноля. Стадия А 2-2-(3-имидазолил)-этил-1,3-диоксолан. К раствору 1,2 г бензимидазола в 15 мл диметилформамида прибавляют 0,49 г гидрида натрия в виде 50 -ной дисперсии в масле. Температура повышается до 35 С, 10 мин спустя после окончания газовыделения прибавляют, позволяя температуре повышаться до 35 С, 1,2 мл 2-(2-бромэтил)-1,3-диоксолана. Осуществляют перемешивание в течение двух часов, затем прибавляют воду, насыщенную хлоридом натрия, после чего экстрагируют простым эфиром, сушат, фильтруют и выпаривают при пониженном давлении. Получают 2 г остатка, который подвергают хроматографии на двуокиси кремния, используя в качестве элюанта метиленхлорид-метанол (95-5). Таким образом получают 1,6 г целевого продукта. ЯМР 3 2,25 и 4,35 группы СН 2 этила 3,85-4,00 группы СН 2 диоксолана 4,87 СН диоксолан 7,29-7,46-7,814 Н бензимидазол 7,92 Н в положении 2 имидазола. Стадия В 3-имидазолилпропаналь. Осуществляют перемешивание в течение 5 ч при рефлюкс раствора 1,6 г продукта, полученного на стадии А, 1,45 г паратолуолсульфокислоты в 60 мл метанола. С помощью карбоната калия устанавливают рН 8,метанол удаляют при пониженном давлении, экстрагирование осуществляют метиленхлоридом, промывают водой, сушат и выпаривают досуха при пониженном давлении. Получают 1,45 г промежуточного диметоксикеталя, который перемешивают при температуре 40 С в течение 18 ч в присутствии 70 мл ацетона и 34 мл 2 соляной кислоты, затем выпаривают ацетон при пониженном давлении, а рН доводят до значения 8-9 с помощью 32 -ного гидрата окиси аммония, осуществляют экстрагирование с помощью метиленхлорида,промывание водой, высушивание и выпаривание досуха при пониженном давлении. Получают 1,13 г продукта, который подвергают хроматографии на двуокиси кремния, используя в качестве элюанта метиленхлорид-метанол 95-5. Получают 0,796 г целевого продукта. ЯМР 3 250 Мгц 3,07- 4,52 группы СН 2 этила 7,25-7,50 ароматические 9,79 СН альдегида. Пример 7. 11,12-дидеокси-3-де-2,6-дидеокси-3-С-метил-3-О-метилрибогексопиранозил)-окси)-6-О-метил-3 оксо-12,11-(оксикарбонил-(2-(3-(2-фенил-5-тиазолил)-пропил)-гидразоно-эритромицин. Осуществляют перемешивание в течение 4 часов при температуре окружающей среды 200 мг продукта,полученного в примере 1, 139 мг 3-(2-фенил-5-тиазолил)-пропаналя (получение которого описано ниже),180 мл уксусной кислоты и 7 мл метанола, после чего добавляют 60 мг цианоборгидрида натрия. Реакционную смесь перемешивают еще в течение 18 ч при температуре окружающей среды, после чего выпаривают насухо при пониженном давлении, остаток поглощают смесью вода-этилацетат, устанавливают значение рН 9 с помощью водного раствора гидрата окиси аммония. Осуществляют экстрагирование этилацетатом, затем промывают в воде, сушат, выпаривают насухо при пониженном давлении. Получают 354 мг продукта, который подвергают хроматографии на двуокиси кремния, используя в качестве элюанта этилацетат, а затем этилацетат-триэтиламин (96-4). Получают 170 мг продукта, который кристаллизуют в смеси этилацетат-пентан, 1-5. Таким образом получают 80 мг целевого продукта. Анализ С 43 Н 64410 829,07 С Н 4361 1 К суспензии 78 г тиобензамида в 200 мл бензола прибавляют раствор формилхлорацетатэтила в 240 мл бензола. Реакционную массу нагревают с рефлюксом в течение 3 ч 30 мин, удаляя образовавшуюся воду. Затем реакционную массу охлаждают и медленно прибавляют 320 мл 20 -ного раствора карбоната калия и 220 мл воды, после чего экстрагируют простым эфиром, промывают, сушат и дистиллируют при пониженном давлении и получают 75,5 г целевого продукта. Стадия В 2-фенил-5-тиазолкарбоновая кислота. К раствору 75,5 г продукта, полученного на стадии А в 130 мл этанола, прибавляют 28,56 г гидроокиси калия в виде таблеток, растворенных в 410 мл этанола, осуществляют нагревание реакционной массы в течение 15 мин, затем охлаждают и отжимают соль калия, промывают в простом эфире и сушат при пониженном давлении. Получают 53,5 г промежуточной соли калия, которую растворяют в 1,2 л воды, подкисляют до рН 1 раствором концентрированной соляной кислоты после фильтрования, получают 29 г целевого продукта рассчитанный 58,52 3,43 6,82 15,6 обнаруженный 58,5 3,7 6,8 15,2. Стадия С 2-фенил-5-тиазолкарбоксилатметил. К раствору 4,77 г кислоты, полученной на стадии В, в 160 мл метанола, прибавляют 2,5 мл ацетилхлорида и осуществляют нагрев при рефлюксе в течение 18 ч. При пониженном давлении доводят состояние реакционной массы до сухого, после чего обрабатывают этилацетатом, фильтруют, концентрируют до уменьшенного объема и отжимают полученные кристаллы. Маточный раствор промывают гидроокисью натрия,экстрагируют этилацетатом, промывают водой и выпаривают досуха, соединяют 2 кристаллизованные фракции и получают 4,54 г целевого продукта (Тпл 108 С). Стадия 2-фенил-5-формилтиазол. Восстановление. К суспензии 1,45 г гидрида литий-алюминия в 65 мл тетрагидрофурана, охлажденной до 10 С, добавляют в течение 20 мин, поддерживая температуру 10 С, раствор 4,5 г продукта, полученного на стадии С, в 35 мл тетрагидрофурана осуществляют перемешивание в течение 45 мин при температуре 10 С, затем в течение 2 ч при температуре окружающей среды. После этого прибавляют тетрагидрофуран, содержащий сначала 10, а затем 50 воды, поддерживая температуру ниже 20 С, после этого прибавляют 15 мл раствора двойной калий-натриевой винно-кислой соли, затем фильтруют, промывают, высушивают насухо при пониженном давлении полученный остаток сгущают в гексане, отжимают и сушат при температуре 40 С при пониженном давлении, получают 3,6 г продукта. Тпл 82 С. Окисление. Осуществляют перемешивание в течение 2 ч 30 мин при температуре окружающей среды 3,57 г продукта,полученного выше, с 143 мл толуола и 17,9 г двуокиси марганца. Реакционную смесь фильтруют и сушат досуха при пониженном давлении. Остаток обрабатывают в гексане, отжимают и сушат при температуре 40 С при пониженном давлении, при этом получают 3,09 г целевого продукта. Тпл 94 С. Стадия Е 3-(2-фенил-5-тиазолил)-пропеналь. В течение 10 минут прибавляют 5 г (формилметилен)-трифенилфосфорана к раствору 2,098 г продукта,полученного на стадии , после чего перемешивают полученную массу в течение 27 ч при температуре окружающей среды. Затем осуществляют выпаривание досуха при пониженном давлении и получают 6,60 г продукта, который подвергают хроматографии на двуокиси кремния, используя в качестве элюата этилацетат-циклогексан (2-8). Получают 1,22 г продукта, который сгущают в пентане с получением 1,047 г целевого продукта (Тпл 104 С). ЯМР 3 (250 Мгц) 8,04 Н триазол 6,49 (, Т 7,5) и 7,69 (, Т 15,5) Н пропен 9,67 (Т 7,5) СНО 7,503 Н и 7,97 2 Н ароматические соединения. Стадия 3-(2-фенил-5-тиазолил)-пропенол. К суспензии 475 мг боргидрида натрия в 50 мл этанола присоединяют порциями 900 мг альдегида, полученного на стадии Е, описанной выше, перемешивают затем в течение 20 мин при температуре окружающей среды, после чего удаляют избыток боргидрида натрия путем прибавления ацетона. Осуществляют выпаривание насухо при пониженном давлении, а затем обрабатывают остаток этилацетатом, промывают солевым раствором, сушат и при пониженном давлении доводят до сухости, при этом получают 960 мг продукта, используемого в том виде как он есть на следующей стадии. Стадия 2-фенил-5-тиазолилропанол. Гидрируют в течение 12 часов при давлении 1 атм., а затем при давлении 1,4 атм. в течение 9 ч раствор 960 мг продукта, полученного на стадии , в 10 мл метанола в присутствии 150 мг палладия на угле. После 4361 1 фильтрования выпаривают насухо при пониженном давлении и остаток подвергают хроматографии на двуокиси кремния (элюант этилацетат-циклогексан, 4-6). Получают 759 мг целевого продукта. ЯМР 3 200 Мгц 1,52 ОН 3,74- 1,97- 2,92 группы 2 7,40-7,905 ароматические 7,53 (,1) Н тиазол. Стадия Н 2-фенил-5-тиазолпропаналь. К раствору, охлажденному до 10 С, 584 мг продукта, полученного на предыдущей стадии, 800 мкл диметилсульфоксида, 1,15 мл триэтиламина и 8 мл метиленхлорида добавляют, поддерживая температуру 10 С,1,27 г комплекса сульфотриоксид-пиридиния. Реакционную массу перемешивают 1 ч 15 мин при температуре 10 С, затем доводят реакционную массу до температуры окружающей среды, после чего осуществляют эктрагирование метиленхлоридом, промывание водой, высушивание и выпаривание насухо при пониженном давлении, в результате получают 806 мг продукта, который подвергают хроматографии на двуокиси кремния(элюант этилацетат-циклогексан, 3-7), при этом получают 450 мг целевого продукта. ЯМР 3 200 Мгц ЯМР 3 200 Мгц 2,88-3,20 группы Н 2 пропил 7,55 Н тиазол 7,403 Н и 7,872 Н Н ароматические 9,85 СНО. Пример 8. 11,12-дидеокси-3-де-2,6-дидеокси-3-С-метил-3-О-метилрибогексопиранозил)-окси)-6-О-метил-3 оксо-12,11-(оксикарбонил-(2-(3-(4-фенил-1 Н-имидазол-1-ил)-пропил)-гидрзоно-эритромицин. В течение 20 ч осуществляют перемешивание 125 мг продукта, полученного в примере 1, 80 мг 3-(4 фенил-1 Н-имидазол-1-ил)-пропаналя (получение которого приведено ниже) и 2 мл метанола. Затем прибавляют 54 мг цианоборгидрида натрия. После этого реакционную массу концентрируют при пониженном давлении, затем обрабатывают 20 мл этилацетата, промывают в гидроокиси натрия, затем водой, насыщенной хлоридом натрия, после чего сушат, выпаривают насухо при пониженном давлении и осуществляют хроматографию остатка на двуокиси кремния (элюант хлороформ-метанол-гидрат окиси аммония, 95-5-0,5), неочищенный продукт поглощают смесью простой эфир-этилацетат, фильтруют, выпаривают досуха и получают 85 мг целевого продукта. Анализ С 43 Н 655 О 10 812,02 С Н рассчитанный 63,6 8,07 8,62 обнаруженный 63,4 8,2 8,3. ЯМР 3 400 Мгц 3,70 Н в положении 11 4,9813 3,86 Н в положении 2 2,26-(СН)3)2 2,63 6-ОСН 3 5,544,27 и 1,97 группы СН 2 пропил 7,3-7,572 Н имидазол 7,2-7,35-7,8 ароматические соединения. К примеру 8 Приготовление 3 -(4-фенил-1 Н-имидазол-1-ил)-пропаналя. Стадия А 3-(4-фенил-1 Н-имидазол-1-ил)-этил-1,3-диоксолан. Действуют в условиях, аналогичных указанным на стадии А получения к примеру 6, однако используют в качестве исходных веществ 1,44 г 4-фенилимидазола и 1,17 мл бромэтилдиоксолана, при этом после хроматографии, осуществленной на двуокиси кремния, получают 1,8 г целевого продукта. ЯМР 3 2,19 (, ) и 4,13 СН 2 пропил 3,8-4,05 группы СН 2 диоксолана 4,88 Н оксолан 7,23 и 7,53 группы СН имидазол 7,23-7,37-7,75 ароматические соединения. Стадия В 3-(4-фенил-1 Н-имидазол-1-ил)-пропаналь. В течение 20 часов нагревают при температуре 60 С 1,77 г продукта, полученного на стадии А, указанной выше, 35 мл ацетона и 30 мл 2 соляной кислоты. Ацетон затем удаляют при пониженном давлении, а раствор нейтрализуют, добавляя в него бикарбонат натрия, после чего осуществляют экстрагирование этилацетатом, высушивание, выпаривание насухо при пониженном давлении. Остаток подвергают хроматографии на двуокиси кремния (элюант этилацетат-метанол, 97-3). Получают 900 мг целевого продукта. ЯМР 3 250 Мгц 9,81 СНО 7,10-7,76 Н имидазол и ароматические 3,01 и 4,29 Н пропил. Пример 9. 11,12-дидеокси-3-де-2,6-дидеокси-3-С-метил-3-О-метилрибогексопиранозил)-окси)-6-О-метил 12,11-(оксикарбонил-(2-(3-(3-фенил-1,2,4-оксодиазол-5-ил)-пропил)-гидразоно)-эритромицин. Действуют в условиях, аналогичным указанным в примере 6, но исходя из 125 мг соединения, полученного в примере 1, и используя 40 мг 3-(3-фенил-1,2,4-оксадиазол-5-ил)-пропаналя (получение которого описано ниже). После хроматографии на двуокиси кремния (элюант простой изопропиловый эфиртриэтиламин-метанол, 90-10-10) и кристаллизации в смеси простого изопропилового эфира метанолом получают 107 мг целевого продукта. 14 пропильные группы СН 2 7,47-8,08 ароматические соединения. К примеру 9 Приготовление 3-(3-фенил-1,2,4-оксадиазол-5-ил-пропаналя. Стадия А 3(3-фенил-1,2,4-оксадиазол-5-ил)-пропанол. В течение 1 ч при температуре окружающей среды перемешивают 2 М раствор 2,5 мл комплекса гидрид бора-метилсульфид в тетрагидрофуране, 920 мг 3-(3-фенил-1,2,4-оксадиазол-5-ил)-пропанойной кислоты(полученной согласно , . , 21, 1193 (1984) и 20 мл тетрагидрофурана. В течение 5 минут прибавляют 10 мл метанола. При пониженном давлении выпаривают досуха, а остаток хроматографируют на двуокиси кремния (элюант этилацетат-гексан, 6-4). Получают 485 мг целевого продукта. ЯМР 3 250 Мгц 2,07 ОН 2,14- 3,10- 3,8 группы СН 2 7,41-7,54-8,06 ароматические соединения. Стадия В 3-(3-фенил-1,2,4-оксадиазол-5-ил)-пропаналь. К раствору, охлажденному до 10 С, 460 мг продукта, полученного на стадии А, 680 мкл диметилсульфоксида и 970 мкл триэтиламина в 5 мл метиленхлорида прибавляют, поддерживая температуру на уровне 10 С, 1,07 г комплекса пиридиний-сульфотриоксид, реакционную массу оставляют до тех пор, пока она не приобретет температуру окружающей среды, затем прибавляют 15 мл метиленхлорида, промывают водой,сушат, выпаривают досуха при пониженном давлении и осуществляют хроматографию на двуокиси кремния(элюант этилацетат-гексан, 4-6), при этом получают 365 мг целевого продукта. ЯМР 3 3,13- 3,26 группы СН 2 7,49-8,05 ароматические соединения. Пример 10. 11,12-дидеокси-3-де-2,6-дидеокси-3-С-метил-3-О-метилрибогексопиранозил)-окси)-6-О-метил-3 оксо-12,11-(оксикарбонил-(2-(3-(2-хлорфенил)-пропил)-гидразоно)-эритромицин. Действуют в условиях, аналогичных указанным в примере 6, но исходят из 125 мг продукта, полученного в примере 1, используя 67 мг 2-хлорфенилпропаналя (получение которого дается ниже). После хроматографии на двуокиси кремния (элюант простой изопропиловый эфир-триэтиламин-метанол, 90-10-10) получают 48 мг целевого продукта. Анализ 4062310 780,40 С Н рассчитанный 61,56 8,01 5,38 4,45 обнаруженный 61,4 8,0 5,4 4,5. ЯМР 3 400 Мгц 3,73 Н в положении 11 5,13 Н в положении 13 3,87 Н в положении 2 2,26-(3)2 2,646-ОСН 3 5,361,83- 2,70-2,79 группы 2 7,05-7,2 ароматические соединения. К примеру 10 Приготовление 3-(2-хлорфенил)-пропаналя. Стадия А 3-(2-хлорфенил)-метилпропаноат. В течение 1 часа в инертной атмосфере перемешивают 4,35 г метахлоркоричной кислоты, 430 мг палладия на угле и 70 мл метанола. Реакционную массу затем перемешивают в течение 3 ч в атмосфере водорода. После этого фильтруют и выпаривают досуха при пониженном давлении, а остаток подвергают хроматографии на двуокиси кремния (элюант этилацетат-гексан, 2-8), получают 3,1 г целевого продукта. ЯМР 3 250 Мгц 2,6- 2,8 группы СН 2 3,6 ОСН 3 7,05-7,37 ароматические соединения. Стадия В 3-(2-хлорфенил)-пропанол К раствору 1,85 г продукта, полученного на стадии А, в 20 мл тетрагидрофурана прибавляют при температуре 0 С 30 мл гидрида диизобутилалюминия в виде 1-раствора в тетрагидрофуране. Реакционную смесь оставляют до тех пор, пока она не приобретет температуру окружающей среды, и перемешивают ее в течение 2 ч. После этого прибавляют смешанную калий-натриевую виннокислую соль, разбавленную тетрагидрофураном, затем фильтруют и выпаривают насухо при пониженном давлении. Остаток подвергают хроматографии на двуокиси кремния (элюант этилацетат-гексан, 2-8), получают 1 г целевого продукта. Стадия С 3-(2-хлорфенил)-пропаналь. Действуют аналогично указанному на стадии В, раздел Приготовление к примеру 9, но исходят из 1 г продукта, полученного на вышеописанной стадии В, с использованием 2,5 мл триэтиламина, 1,75 мл диме 15 4361 1 тилсульфоксида и 2,8 г комплекса пиридиний-сульфотриоксид. После хроматографии на двуокиси кремния при использовании в качестве элюанта этилацетат-гексана (1-9) получают 425 мг (43 ) целевого продукта. ЯМР 3 250 Мгц. 2,79 и 2,94 группы СН 2 7,05-7,25 ароматические соединения 9,82 СНО. Пример 11. 11,12-дидеокси-3-де 2,6-дидеокси-3-С-метил-3-О-метилрибогексопиранозил)-окси)-6-О-метил-3 оксо-12,11-(оксикарбонил-(2-(3-(4-хинолинил)-пропил)-гидразоно-эроитромицин. Стадия А 11,12-дидеокси-3-де-2,6-дидеокси-3-С-метил-3-О-метилрибогексопиранозил)-окси)-6-О-метил-3 оксо-12,11-(оксикарбонил-(гидразоно)-эритромицин, изомер 10 и изомер 10 соответственно. В 176 мл метилцианида растворяют 17,65 г 2-ацетат-11-деокси-10,11-дидегидро-3-де-(2,6-дидеокси-3-Сметил-3-О-метилрибогексопианозил)-окси)-12-О-(1 Н-имидазол-1-ил)-карбонил)-6-О-метил-3-оксоэритромицина. Прибавляют 4,07 г карбоната цезия и 25,5 гидразингидрата. Нагревают 10 мин при температуре 85 С, отгоняют растворитель при пониженном давлении и температуре, равной 40 С, экстрагируют метиленхлоридом, промывают водой, сушат, выпаривают растворитель, остаток поглощают метанолом, отжимают выпавший осадок и сушат его при температуре 50 С при пониженном давлении, получают 6,04 г продукта. Концентрируют досуха маточные воды, осуществляют хроматографию на двуокиси кремния (элюант простой изопропиловый эфир-метанол-триэтиламин, 80-10-10) и получают 0,83 г изомера(0,4) и 2,65 г изомера В (0,2). Стадия В 11,12-дидеокси-3-де-2,6-дидеокси-3-С-метил-3-О-метилрибогексопиранозил)-окси)-6-О-метил-3-оксо-12,11-(оксокарбонил-(2-(3-(4-хинолинил)-пропил)-гидразоно-эритромицин. В 130 мл метанола суспендируют 13 г продукта, полученного на стадии А, и 4,66 г 4-хинолинпропаналя,полученного как указано ниже. Прибавляют 4,8 мл уксусной кислоты и перемешивают в течение 20 ч при температуре окружающей среды. Затем добавляют 5,3 г цианоборгидрида натрия, после чего продолжают перемешивание в течение 4 ч. Удаляют метанол при пониженном давлении, экстрагируют этилацетатом,промывают водным -раствором гидрата окиси натрия, а затем водой выпаривают растворитель из органической фазы, остаток хроматографируют на двуокиси кремния (элюант этилацетат-триэтиламин, 97-3) и получают 12,7 г продукта с 0,15. После новой хроматографии на двуокиси кремния (элюант метиленхлорид- метанол, 95-5, затем 85-15) и кристаллизации в простом изопропиловом эфире получают чистый продукт, Тпл 183 С, данные анализа которого идентичны данным анализа продукта из примера 5. К примеру 11 Приготовление 4-хинолинпропаналя. Стадия А 2-(4-хинолинэтенил)-1,3-диоксолан. В 40 мл тетрагидрофурана суспендируют 3,15 г 4-хинолинкарбоксальдегида и 8,6 г 1,3-(диоксалан-2 ил)метил-трифенилфосфонийбромида, охлаждают ее до температуры -30 С, затем прибавляют 2,5 г трет.бутилата калия и перемешивают в течение 1 ч. Реакционную массу оставляют до тех пор, пока она не приобретет температуру окружающей среды, затем перемешивают в течение 3 ч, выливают на смесь вода/лед, экстрагируют метиленхлоридом, промывают водой, сушат, выпаривают растворитель при пониженном давлении, остаток поглощают смесью простой этиловый эфир- пентан,3-7, перемешивают в течение 2 ч,фильтруют и выпаривают растворитель фильтрата, получают 3,99 г желаемого продукта. Стадия В 2-2-)4-хинолинил)-этил-1,3-диоксолан. В 40 г метанола растворяют 4,3 г продукта, полученного на стадии А, прибавляют 0,215 г активированного угля с 10 палладия и гидрируют в течение 2 ч под давлением 1500 мбар. Фильтруют, промывают в метаноле, выпаривают растворитель и получают 4,2 г целевого продукта, используемого в том виде, как он есть на последующей стадии. Стадия С 4-хинолинпропаналь. 4,2 г продукта, полученного на стадии В, растворяют в 70 мл ацетона, после чего прибавляют 70 мл 2 соляной кислоты. Реакционную смесь нагревают в течение 6 ч при температуре 40 С, удаляют ацетон при пониженном давлении, экстрагируют этилацетатом, промывают водой, затем с помощью водного раствора гидрата окиси аммония доводят рН водной фазы до значения 9. Экстрагируют этилацетатом, объединяют органические фазы, сушат их и выпаривают растворитель. После хроматографии на двуокиси кремния (элюант этилацетат-циклогексан, 6-4) получают 1,36 г целевого продукта. Пример 12. 11,12-дидеокси-3-де 2,6-дидеокси-3-С-метил-3-О-метилрибогесопианозил)-окси)-6-О-метил-3-оксо 12,11-(оксикарбонил-2-(3-(7-метокси-4-хиролинил)-пропил)-гидразоно-эритромицин. Растворяют в 2 мл метанола 299 мг 7-метокси-4-хинолинпропаналя, получение которого указано ниже, и 313,9 мг продукта А, полученного в примере 1, и 120 мкл уксусной кислоты. Перемешивают 2 ч 15 мин при температуре окружающей среды, затем прибавляют 62,84 мг цианоборгидрида натрия. Перемешивают в течение 20 ч при температуре окружающей среды. Реакционную массу выливают на 50 мл этилацетата, промывают в 15 мл -гидрата окиси натрия, затем в воде, сушат, выпаривают растворитель при пониженном давлении и получают 549 мг продукта, который очищают путем хроматографии на двуокиси кремния (элю 16 4361 1 ант простой изопропиловый эфир- метанол-триэтиламин, 80-10-10, затем хлороформ-метанол-гидрат окиси аммония, 96-4-0,4). Получают 37,2 мг целевого продукта,0,2. Анализ С Н 2 2,656-ОСН 3 2,658 0,82 СН 3 СН 2. К примеру 12 Приготовление 7-метокси-4-хинолинпропаналя. Стадия А 2-(7-метокси-4-хинолинил)-этенил-1,3-диоксолан. Работают, как указано в приготовлении к примеру 11, стадия А, но используют в качестве исходного соединения 787 мг 7-метокси-4-хинолинкарбоксальдегида. Получают 2,61 г продукта, который подвергают хроматографии на двуокиси кремния (элюант хлороформ-этилацетат, 7-3). Получают 931 мг целевого продукта. Стадия В 2-2-(7-метокси-4-хинолинил)-этил-1,3-диоксолан. Работают, как указано в приготовлении к примеру 11, стадия В, используя 931 мг продукта, полученного на стадии А, и получают 869 мг целевого продукта. Стадия С 2-(7-метокси-4-хинолин)-пропаналь. Работают, как указано в приготовлении к примеру 11, стадия С, но используют 845 мг продукта, полученного на стадии В. Получают 310 мг целевого продукта,0,15. Пример 13. 11,12-дидеокси-3-де-2,6-дидеокси-3-С-метил-3-О-метилрибогексопиранозил)-окси)-6-О-метил-3 оксо-12,11-(оксикарбонил-2-(3-(3-пиридинил-4-тиазолил)-пропил)-гидразоно-эритромицин. Процесс получения вышеназванного соединения аналогичен указанному в примере 12, однако в качестве исходного продукта используют взятые в 3,7 г метанола 158 мг 2-(3-пиридинил)-4-тиазолпропаналя, 370 мг соединения А, полученного в примере 1, и 70 мкл уксусной кислоты, а затем после перемешивания в течение 4 ч при температуре окружающей среды, 75 мг цианоборгидрида натрия. После перемешивания реакционной среды в течение 16 ч при температуре окружающей среды, снова прибавляют 16 мг альдегида и 20 мг восстанавливающего реагента, после чего продолжают перемешивание еще в течение 3 ч. Затем прибавляют воду, этилацетат, подщелачивают реакционную массу до рН 9 с помощью гидрата окиси аммония, промывают органическую фазу водой, сушат и выпаривают растворитель при пониженном давлении. После хроматографии на двуокиси кремния (элюант простой изопропиловый эфир-метанол-триэтиламин, 80-10-10) получают 203 мг целевого продукта. 3,1810 3,7411 7,05 Н 5 тиазол 7,37- 8,24 ( - 8,62- 9,13 пиридин 3,862 2,656-ОСН 3 2,668 0,8532. К примеру 13 Приготовление 2-(3-пиридинил)-4-тиазолпропаналя. Стадия А 2-(3-пиридинил)-4-тиазолил этенил-1,3-диоксолан. Работают, как в приготовлении к примеру 11, стадия А, однако используют в качестве исходного вещества 2,6 г 2-(3-пиридинил)-4-тиазолкарбоксальдегида. После хроматографии, осуществленной на двуокиси кремния (элюант этилацетат-гексан, 2-10 получают 4,8 г целевого продукта (0,35), используя в том виде,как он есть на последующей стадии. Стадия В 2-2-3-пиридинил)-4-тиазолил)-этил-1,3-диоксолан. Работают, как указано в приготовлении к примеру 11, стадия В, но используют в качестве исходного вещества 4,8 г продукта, полученного на стадии А. После хроматографии остатка, осуществленной на двуокиси кремния (элюант этилацетат-циклогексан, 2-1), получают 1,4 г целевого продукта. Стадия С 2-(3-пиридинил)-4-тиазолилпропаналь. Работают, как указано в приготовлении к примеру 11, стадия С, но используют в качестве исходного вещества 1,2 г продукта, полученного на стадии В. После хроматографии, осуществленной на двуокиси кремния (элюант этилацетат-гексан, 2-1), получают 468 мг целевого продукта. Действуя аналогично указанному в предыдущих примерах, используя в качестве исходного вещества соединение, согласно примеру 1, и соответствующий альдегид, получают следующие продукты. Пример 14. 11,12-дидеокси-3-де-2,6-дидеокси-3-с-метил-3-О-метилрибогексопиранозил)-окси)-6-О-метил-3 оксо-12,11-(оксикарбонил-2-(3-(1 Р-имидазол-1-ил)-пропил)-гидразоно-эритромицин. ЯМР 3 300 Мгц 0,83 СН 3-СН 2 1,08 -1,17- 1,25- 1,3- 1,35 группа СН 3-СН 1,3- 1,476 и 122,122-2-СН 2 2,272 2,45 Н 3 2,596-ОМе 3,054 2,6-3,2 Н 2, Н 10 8 и Н 2 3,53 Н 5 3,7211 3,852 4,27 1, и 5 4,632- 4,9913 5,462 7,10-7,64-7,66-7,97 ароматические соединения. 8 3,76 Н 11 3,852 5,06 Н 13 5,39-2 7,49-7,94 ароматические соединения. Пример 19. 11,12-дидеокси-3-де-2,6-дидеокси-3-С-метил-3-О-метилрибогексопиранозил)-окси)-6-О-метил-3 оксо-12,11-(оксикарбонил-2-(3-(4-хлорфенил-1 Н-имидазол-1-ил)-пропил)-гидразоно-эритромицин. Пример 20. 11,12-дидеокси-3-де-(2,6-дидеокси-3-С-метил-3-О-метилрибогексопиранозил)-окси)-6-О-метил-3 оксо-12,11-(оксикарбонил-(2-(3-(6-метокси-4-хинолинил)-пропил)-гидразоно-эритромицин. Действуя аналогично указанному в примере 12, получают целевой продукт. ЯМР 3 300 Мгц 3,7411 552-2 3,98 ОСН 3 хинолина 7,25-7,35- 7,99- 8,65 Н хинолин 3,87 Н 2 2,646-ОСН 3 2,648 5,0213. Действуя аналогично указанному выше, получают соединения формулы , где радикал представляет собой радикал. Пример фармацевтической композиции. приготавливают таблетки, содержащие продукт, полученный в примере 5 150 мг эксципиент (в количестве, достаточном для) 1 г. Описание эксципиента крахмал, тальк, стеарат магния. Фармакологическое исследование заявляемых соединений. Метод разбавления в жидкой среде. Приготавливают серию пробирок, в которые помещено одинаковое количество стерильной питательной среды. Вводят в каждую пробирку возрастающее количество исследуемого соединения, затем каждую пробирку обсеменяют штаммом бактерий. После инкубации в течение двадцати четырех часов в термостате при 37 С торможение роста оценено с помощью просвечивания, что позволяет определить минимальные ингибирующие концентрации (МИК), выраженные в микрограммах/см 3. 18 4361 1 Нижеследующие результаты получены при использовании продукта из примера 5 (по истечении 24 ч). Грамположительные штаммы бактерий 011 С 4 0,020110251 0,08012011 0,04 группа 02110,02 группа В 021 НТ 10,02 группа 02210,02 группа 02310,02 группа 02050,020210,02 группа В 02110,0203210,0203050,02. Более того, соединение, полученное в примере 5, продемонстрировало интересную активность в отношении грамотрицательных штаммов бактерий 351 НТ 3, 351 СА 1 и 3516. Соединения, полученные в примерах 12 и 13 также проявили прекрасную активность в отношении грамположительных и грамотрицательных штаммов бактерий. Действуя аналогично указанному выше, были получены следующие результаты при использовании соединений из примеров 12 и 13 (по истечении 24 ч). Грамположительные штаммы бактерий Пример 12 Пример 130114 0,08 0,04011025 0,08 0,15012011 0,04 0,0402110,020,0202110,020,0202210,020,020231 0,020,020321 0,040,02030510,020,0203018 0,6 0,63513 1,2 0,635112 1,2 1,2. Национальный центр интеллектуальной собственности. 220072, г. Минск, проспект Ф. Скорины, 66. 19
МПК / Метки
МПК: A61P 31/04, C07H 17/08, A61K 31/70
Метки: основе, фармацевтическая, производные, способ, получения, эритромицина, композиция
Код ссылки
<a href="https://by.patents.su/19-4361-proizvodnye-eritromicina-sposob-ih-polucheniya-i-farmacevticheskaya-kompoziciya-na-ih-osnove.html" rel="bookmark" title="База патентов Беларуси">Производные эритромицина, способ их получения и фармацевтическая композиция на их основе</a>
Производные пирролзамещенных уреидов, способ их получения и фармацевтическая композиция на их основе

Номер патента: 2226
Опубликовано: 30.09.1998
Авторы: Марина ЧЬОМЕИ, Мариа Гранди, Джованни Бьясоли, Никола МОНДЖЕЛЛИ, Альфредо Пайо
МПК: A61K 31/40, C07D 207/34, C07D 207/335...
Метки: композиция, фармацевтическая, способ, уреидов, основе, получения, пирролзамещенных, производные
Текст:
...этилолеат, гликоли, такие как пропиленгликоль, и по желанию, соответствующее количество гидрохлорида лидокаина. Лекарственные средства для наружного применения могут быть изготовлены в виде кремов, примочек или паст для кожной обработки, например путем смешивания активного ингредиента со стандартными масляными или эмульгирующими наполнителями. Лекарственные средства для перорального введения могут быть изготовлены в виде таблеток и...
9а-N-(N’-Карбамоил) – или 9а-N-(N’-тиокарбамоил) производные 9-деоксо-9а-аза-9а-гомоэритромицина А, способ их получения, фармацевтическая композиция на их основе

Номер патента: 4304
Опубликовано: 30.03.2002
Авторы: Неделько КУЮНДЖИЧ, Желько КЕЛНЕРИЧ, Габриела КОБРЕХЕЛ
МПК: C07H 17/00, A61K 31/70, A61P 31/04...
Метки: фармацевтическая, получения, способ, производные, 9-деоксо-9а-аза-9а-гомоэритромицина, или, 9а-n-(n’-тиокарбамоил, композиция, 9а-n-(n’-карбамоил, основе
Текст:
...а(4-метил-5-оксазол)-карбамоил-9 а-аза-9 а-гомоэритромицин А (5,4 г, 93,3 ), точка плавления 174177 С. С помощью перекристаллизации из горячего ацетона получали хроматографически гомогенный продукт, имеющий следующие физико-химические постоянные Точка плавления - 181-183 С, -(-6-6)-2(10010020),0,149. СНС 3-СН 3 ОН-конц.Н 4 Н(610,1),0,491 см-1 1730, 1680, 1655, 1490, 1460, 1380, 1170, 1050, 755, 660. 1 9,02(9), 7,95(-), 5,71(1, Н-13),...
Производные фосфолипидов, способ их получения, фармацевтическая композиция на их основе, способ получения фармацевтической композиции

Номер патента: 3733
Опубликовано: 30.12.2000
Авторы: Бернхард Кучер, Юрий Штекар, Юрген Энгель, Герхард Несснер, Петер Хилгард
МПК: C07F 9/09, C07F 9/10, A61K 31/685...
Метки: производные, композиция, способ, композиции, основе, фосфолипидов, получения, фармацевтической, фармацевтическая
Текст:
...например, с карбонатами щелочных и щелочноземельных металлов и органическими аминами. В качестве растворителей служат алифатические спирты, например метанол, этанол и изопропанол. Можно работать при повышенной температуре. При выполнении всех реакций нужно соблюдать обычное в химии металлоорганических веществ исключение влаги и кислорода воздуха. Все четыре способа можно выполнять с проведением дополнительной стадии очистки. Очистку...
Производные 3,4-дигидроизохинолина или их фармацевтически приемлемые соли, способ их получения и фармацевтическая композиция на их основе
Номер патента: 2682
Опубликовано: 30.03.1999
Авторы: Вальтер Лёзель, Отто Роос, Дитрих Арндтс, Ильзе Штреллер, Франц Йозеф Кун
МПК: A61K 31/47, C07D 217/18
Метки: фармацевтическая, фармацевтически, соли, производные, приемлемые, композиция, способ, 3,4-дигидроизохинолина, основе, получения, или
Текст:
...разрушаются и при этом выделяются медиаторы, поражающие ткани, в том числе также и хемотактические (например, АТФ, лейкотриены, простагландины, тринуклеотиди др.). Привлеченные хемотаксисом дополнительные лейкоциты увеличивают зону поражения. Описанные выше опыты показали, что этот порочный круг можно блокировать дачей описанных выше активных веществ. Нейрологическое нарушение остается таким образом ограниченным. Можно было показать, что...
Производные N-(4-пиперидинил) (дигидробензофуран или дигидро-2H-бензопиран)карбоксиамида, способ их получения, фармацевтическая композиция на их основе, промежуточные соединения

Номер патента: 4186
Опубликовано: 30.12.2001
Авторы: ДЕ КЛЕЙН, Михел Анна Йозеф, ВАН ДАЛЕ, Георгес Хенри Паул, БОСМАНС, Ян-Паул Рене Мари Андре
МПК: A61K 31/4523, A61P 1/14, A61K 31/16...
Метки: или, дигидро-2h-бензопиран)карбоксиамида, композиция, дигидробензофуран, получения, производные, n-(4-пиперидинил, способ, основе, соединения, промежуточные, фармацевтическая
Текст:
...гидроксидов, адкоксидов, гидридов, амидов или оксидов (карбонат натрия, гидрокарбонат натрия, карбонат калия, гидроксид натрия, метоксид натрия, гидрид натрия, амид натрия, карбонат кальция, гидроксид кальция, оксид кальция и др.), или неорганические основания, например третичный амин (,-диэтилэтанамин, -/1-метилэтил/-2-пропанамин, 4 этилморфолин и др.), чтобы нужная кислота нейтрализовалась в процессе реакции. В некоторых случаях...
Предыдущий патент: Производные 3-арилоксикарбоновой кислоты, гербицидный препарат и средство для подавления роста растений
Следующий патент: Соединения, селективно ингибирующие ароматазу, способ их получения, фармацевтическая композиция на их основе
Случайный патент: Способ прогнозирования исхода лучевого лечения неоперабельного рака шейки матки