Антагонисты LHRH с улучшенной растворимостью
Номер патента: 7934
Опубликовано: 30.04.2006
Авторы: БЕРНД Майкл, КУТШЕР, Бернхард, БЕКЕРС Томас, РОМАЙС, Петер, ГЮНТЕР, Экхард, РАЙССМАНН, Томас







Текст
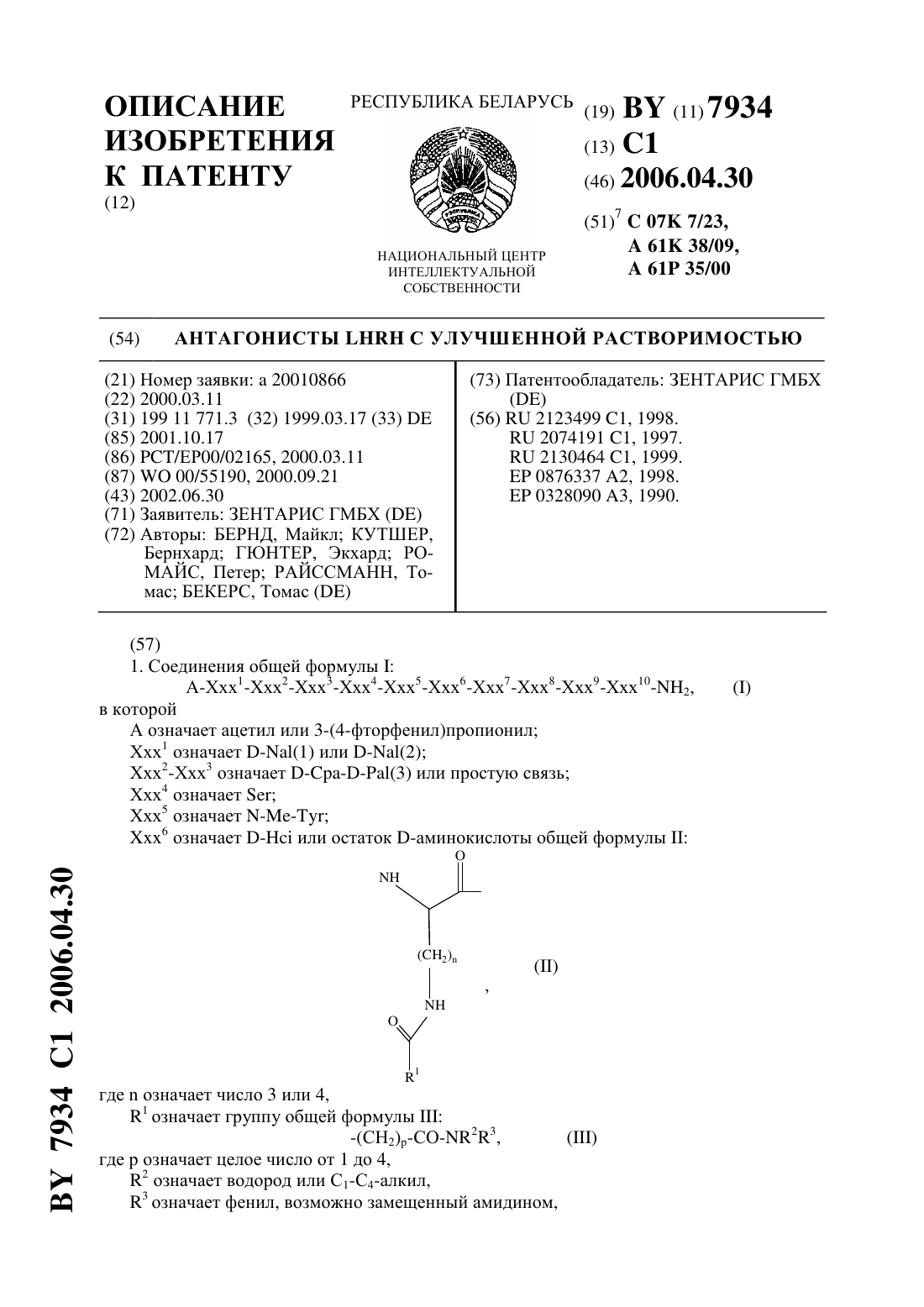
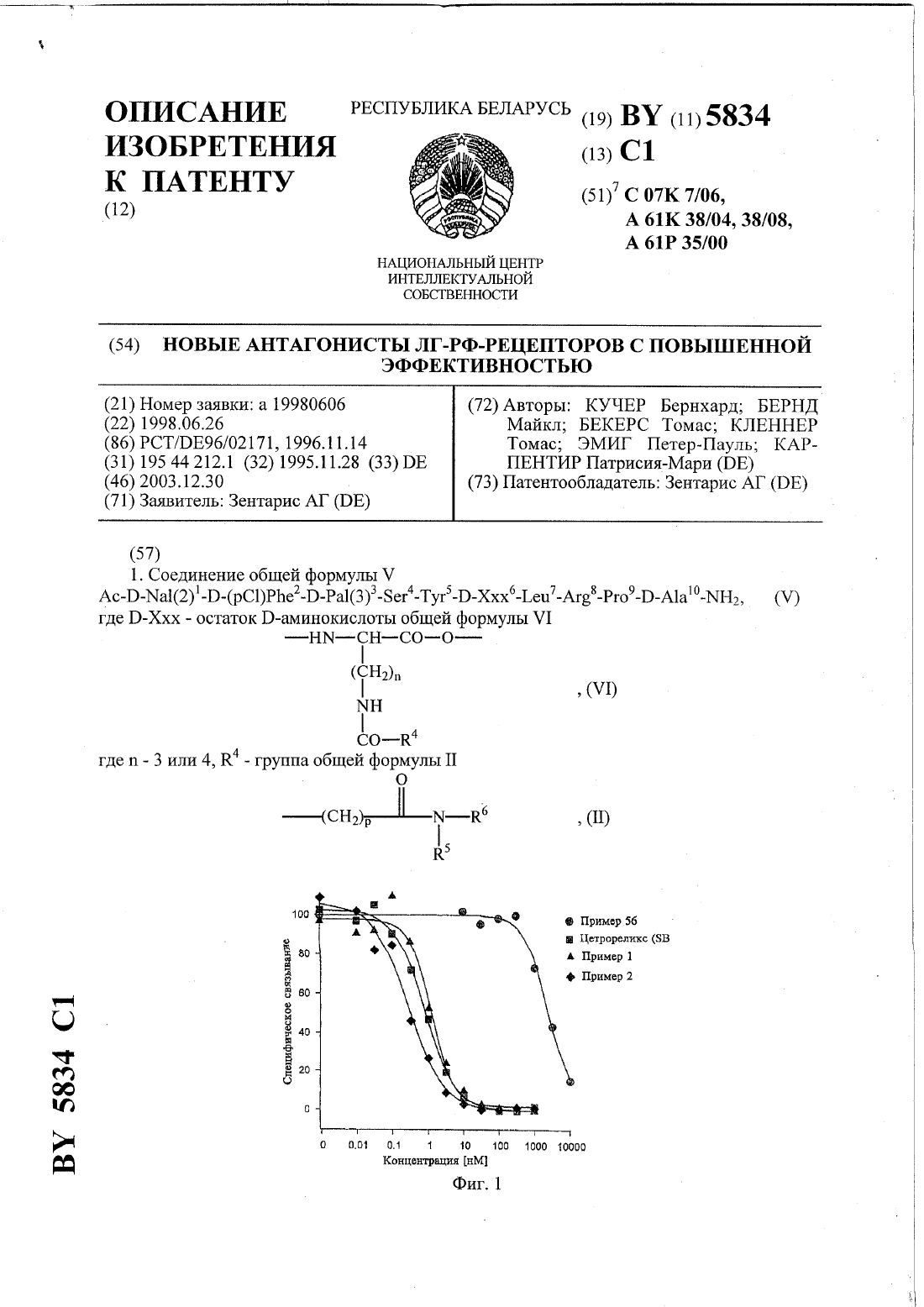
(57) 1. Соединения общей формулыА-Ххх 1-Ххх 2-Ххх 3-Ххх 4-Ххх 5-Ххх 6-Ххх 7-Ххх 8-Ххх 9-Ххх 10-2,в которой А означает ацетил или 3-(4-фторфенил)пропионил Ххх 1 означает -(1) или -(2) Ххх 2-Ххх 3 означает (3) или простую связь Ххх 4 означаетХхх 5 означает -6 означает - или остаток -аминокислоты общей формулы гдеозначает число 3 или 4,1 означает группу общей формулы-(СН 2)р-СО-23, где р означает целое число от 1 до 4,2 означает водород или С 1-С 4-алкил,3 означает фенил, возможно замещенный амидином,7934 1 2006.04.30 или 1 означает 3-амино-1,2,4-триазол-5-карбонильную группу Ххх 7 означаетилиХхх 8 означаетилиХхх 9 означаетХхх 10 означает А 1 а или ,или их соли с фармацевтически приемлемыми кислотами. 2. Соединения по п. 1, отличающиеся тем, что соль представляет собой ацетат, трифторацетат или эмбонат. 3. Соединения по п. 1 или 2, отличающиеся тем, что Ххх 6 означает -(3-амино-1,2,4 триазол-5-карбонил)-, сокращенно -, или 4-(4-амидинофенил)амино 1,4-диоксобутил-, сокращенно -. 4. Соединение по п. 1, отличающееся тем, что представлено формулой (2)1-2(3)3-456-7-8-910-2. 5. Соединение по п. 1, отличающееся тем, что представлено формулой (2)1-2(3)3-456-7-8-910-2. 6. Соединение по п. 1, отличающееся тем, что представлено формулой (2)1-2(3)3-45(В)6-7-8-910-2. 7. Соединение по п. 1, отличающееся тем, что представлено формулой (2)1-2(3)3-456-7-8-910-2. 8. Соединение по п. 1, отличающееся тем, что представлено формулой (2)1-2(3)3-456-7-8-910-2. 9. Соединение по п. 1, отличающееся тем, что представлено формулой (2)1-2(3)3-456-7-8-9-10-2. 10. Соединение по п. 1, отличающееся тем, что представлено формулой (2)1-2(3)3-456-7-8-9-10-2. 11. Соединение по п. 1, отличающееся тем, что представлено формулой 3-(4 фторфенил)пропионил(1)1-456-7-8-910-2. 12. Фармацевтическая композиция, обладающая активностью против гормонзависимых опухолей, а также незлокачественных образований, для лечения которых требуется подавление гормона , включающая соединение по любому из пп. 1-11 в эффективном количестве и фармацевтически приемлемый носитель. 13. Способ получения соединений общей формулыпо п. 1, в котором фрагменты, состоящие из остатковс подходящими защитными группами, гдеозначает целое число от 1 до 10, а Ххх 1 ацетилирован, наращивают на твердой фазе или в растворе в общепринятых условиях, затем фрагменты на твердой фазе соединяют путем конденсации фрагментов и после конденсации соединения общей формулыотщепляют от твердой фазы посредством амидирования по остатку Ххх 10. 14. Применение соединений по пп. 1-11 для получения фармакологического средства для лечения гормонзависимых опухолей, прежде всего карциномы предстательной железы или рака молочной железы, а также для лечения незлокачественных образований, для лечения которых требуется подавление гормона . 15. Способ получения фармакологического средства, в котором соединения по пп. 111 смешивают с общепринятыми носителями или вспомогательными материалами и изготавливают в виде фармакологического средства. Изобретение относится к антагонистам - с улучшенной растворимостью, способам получения этих соединений, лекарственным средствам, в которых содержатся указанные соединения, а также к применению лекарственного средства при лечении гормонзависимых опухолей и незлокачественных гормонзависимых заболеваний, таких как доброкачественная гиперплазия простаты (ВРН) и эндометриоз. Номенклатура, используемая при 2 7934 1 2006.04.30 описании пептидов, совпадает с номенклатурой, которая вошла в состав Биохимической Номенклатуры, утвержденной комиссией ИЮПАК-ИЮБ ( (1984), т. 138, стр. 8-37), где в соответствии с общепринятым написанием -концевая аминогруппа располагается слева, а С-концевая карбоксильная группа располагается справа. Антагонисты -, так же как и пептиды по настоящему изобретению, включают как природные,так и синтетические аминокислоты, причем первые включают , , , , , , , , , , , , , , ,и . Сокращенные названия отдельных аминокислотных остатков основаны на тривиальных названиях аминокислот, например,аланин,аргинин,глицин,лейцин,лизин, (3)3(3-пиридил)аланин, (2)3-(2-нафтил)аланин),фенилаланин, Сра 4-хлорфенилаланин,пролин,серин,треонин, Тгртриптофан, Тутирозин исаркозин. Если не указано иное, все описанные в заявке аминокислоты относятся к-ряду. Например, сокращенное название -(2) означает 3-(2-нафтил)аланин, а сокращенное названиеозначает -серин. Заместители по -аминогруппе боковой цепи лизина приведены после терминав скобках, по выбору в сокращенной форме. Прочие сокращения, использованные в тексте заявки Ас - ацетил- 3-амино-1,2,4-триазол-5-карбонил В - 4-(4-амидинофенил)амино-1,4-диоксобутил Вос - трет-бутилоксикарбонил Вор - гексафторфосфат бензотриазол-1-окситрис(диметил-амино)фосфония- дициклогексилкарбодиимид ДХМ - дихлорметан- диметоксифенилдиметилметиленоксикарбонил (диметиоксидиметил-)- диизопропилкарбодиимид- 1-гидроксибензотриазол ВЭЖХ - высокоэффективная жидкостная хроматография- метил ТФУ - трифторуксусная кислота- бензилоксикарбонил- гомоцитруллин Сра - 4-хлорфенилаланин. Пептиды по настоящему изобретению представляют собой аналоги релизинг-фактора лютеинизирующего гормона (-), как показано на следующей структуре 2, (-, гонадорелин). В течение более, чем 20 лет исследованы селективные потенциальные антагонисты декапептида - (статья.,, 1986. - Т. 7. - С. 44-66). Большой интерес к таким антагонистам обусловлен необходимостью их использования в области эндокринологии, гинекологии, предупреждения беременности и терапии онкологических заболеваний. В качестве потенциальных антагонистов - получено множество соединений. Наиболее интересными соединениями, которые найдены к настоящему времени, являются такие соединения, структура которых является модификацией структуры -. Первую серию потенциальных антагонистов получали введением ароматических аминокислот в положения 1, 2, 3, и 6 или 2, 3 и 6 (полипептидной цепи). Принятый способ описания таких соединений заключается в следующем прежде всего указывают амино 3 7934 1 2006.04.30 кислоты, на которые заменены первоначальные аминокислоты в пептидной цепи -,причем положения, в которых произошли замены, обозначают цифрами, расположенными в верхнем индексе. Затем указывают термин -, обозначающий тот факт, что речь идет об аналоге -, содержащем указанные замены. Известными антагонистами являются следующие соединения 1,2, -3,6 - (статья .,6(Пептиды, Материалы 6-го американского симпозиума по пептидам), стр. 775-779, ред..,.,,(1979)(1983), т. 56, стр. 420). Для усиления действия антагонистов позднее в положение 6 стали вводить основные аминокислоты, например, -. Например, получены 1,2, -3, -6, 10- (-30276) (статья .,(1982), т. 100, стр. 1445), и. ,,, , , (1984. Прочие потенциальные антагонисты - описаны в заявках 92/19651,94/19370,92/17025,94/14841,94/13313, - 5300492, - 5140009,0413209 1 и 19544212 1. Позднее описаны соединения с модифицированными остатками орнитина и лизина в положении 6, представленные следующей формулой (2)12(3)3-456-7-8-910-2, где - является аминокислотным остатком общей формулы Кроме того, антогонисты - описаны в заявках 97/19953 ЕР-А 20328090. Другими известными антагонистами - являются антареликс, ганиреликс и цетрореликс. Антареликс(2)12(3)3-4-56-7-8-910-2. Задачей изобретения является разработка новых антагонистов -, обладающих повышенной устойчивостью к действию ферментов и значительно более высокой растворимостью в воде. Объектом изобретения являются соединения общей формулыА-Ххх 1-Ххх 2-Ххх 3-Ххх 4- Ххх 5-Ххх 6-Ххх 7- Ххх 8-Ххх 9-Ххх 10- Н 2, где А означает ацетил- или 3-(4-фторфенил)пропионилгруппу,Ххх 1 означает -(1) или -(2),Ххх 2-Ххх 3 означает (3) или простую связь,Ххх 4 означает ,4 гдеозначает число 3 или 4, причем 1 означает группу общей формулы-(2)23, где р означает целое число от 1 до 4, 2 означает водород или С 1-С 4-алкил,3 означает фенил, возможно замещенный амидином, или 1 означает 3-амино-1,2,4-триазол-5 карбонильную группу Ххх 7 означаетили ,Ххх 8 означаетили ,Ххх 9 означает , и Ххх 10 означаетили ,или их соли с фармацевтически приемлемыми кислотами, прежде всего ацетата, эмбоната и трифторацета. Среди соединений по изобретению более предпочтительными являются соединения, в которых Ххх 6 означает -(3-амино-1,2,4-триазол-5-карбонил)-, сокращенное название-. Другими более предпочтительными соединениями по изобретению являются 3-(4-фторфенил)пропионил-О-а(1)1-4 Ме-56-7-А 8-9- -2,а также их соли с вышеупомянутыми фармацевтически приемлемыми кислотами. Другим объектом изобретения является фармацевтическая композиция, обладающая активностью против гормонзависимых опухолей, а также незлокачественных образований,для лечения которых требуется подавление гормона -, включающая соединение по изобретению в эффективном количестве и фармацевтически приемлимый носитель. Еще одним объектом изобретения является способ получения соединений общей формулы , в котором фрагменты, состоящие из остатков Ххх с подходящими защитными группами, гдеозначает целое число от 1 до 10, а Ххх 1 ацетилирован, наращивают на 10 7934 1 2006.04.30 твердой фазе или в растворе в общепринятых условиях, затем фрагменты на твердой фазе соединяют путем конденсации фрагментов и после конденсации соединения общей формулыотщепляют от твердой фазы посредством амидирования по остатку Ххх 10. Объектом изобретения является также применение соединений по изобретению для получения фармакологического средства для лечения гормонзависимых опухолей, прежде всего карциномы предстательной железы или рака молочной железы, а также при незлокачественных показаниях, при терапии которых требуется подавление -. С этой целью их смешивают с общепринятыми носителями и вспомогательными веществами и изготавливают в виде лекарственных препаратов. Синтез соединений формулыможно осуществлять как с помощью классической конденсации фрагментов, так и твердофазным синтезом по Мерифильду с последовательным наращиванием цепи с использованием -лизина, ацилированного в боковой цепи карбоновой кислотой общей формулы 1-, а также посредством модификации декапептидного фрагмента соответствующими карбоновыми кислотами за счет образования амидной связи с боковой цепью -лизина 6. Благодаря этому введение 1-СО-группы можно осуществлять на трех различных стадиях синтеза перед конденсацией отдельных фрагментов с образованием пептида, после введения в цепь лизина или орнитина, но перед конденсацией последующих фрагментов, или после конденсации всех фрагментов. Соединения формулысинтезируют известными методами, например, с помощью исключительно твердофазного метода, с частичным использованием твердофазного метода (так называемой конденсацией фрагментов) или с помощью классического синтеза в растворе см. в книгеМ.(Принципы пептидного синтеза),, 1984. Например, методы твердофазного синтеза описаны в руководстве,(Твердофазный синтез пептидов),. , 1984 и в книге.,(Пептиды). Гл.1 1979. С. 1-285.. Классический синтез в растворе подробно описан в руководстве(-),(Методы органической химии (Губен-Вейль), синтез пептидов), ред.., 1974,, , . Ступенчатый синтез проводят, например, следующим образом, прежде всего С-концевую аминокислоту с защищенной -аминогруппой ковалентно присоединяют к одному из обычных нерастворимых носителей, затем от этой аминокислоты отщепляют -аминозащитную группу, и к образовавшейся свободной аминогруппе присоединяют по карбоксильной группе следующую аминокислоту с защищенной аминогруппой, и таким образом к синтезируемому пептиду стадия за стадией присоединяют в правильной последовательности остальные аминокислоты, а после присоединения всех аминокислот готовый пептид отщепляют от носителя и по выбору отщепляют остальные присутствующие защитные группы боковых цепей аминокислот. Ступенчатую конденсацию проводят обычным способом из соответствующих, должным образом защищенных аминокислот. Конденсацию отдельных аминокислот друг с другом проводят обычными методами,которые прежде всего включают метод с использованием симметричного ангидрида в присутствии дициклогексилкарбодиимида или диизопропилкарбодиимида (, ),общий карбодиимидный метод,карбодиимид-гидроксибензотризольный метод (см. в книге(Пептиды). Т. 2. / Под. ред. Е. , . ). При конденсации фрагментов предпочтительно используют не сопровождающийся рацемизацией азидный метод или метод с использованием -1-гидроксибензотриазола или, соответственно, -3-гидрокси-4-оксо-3,4-дигидро-1,2,3-бензотриазина. Кроме того, можно использовать активированный эфир фрагмента. 6 7934 1 2006.04.30 Для ступенчатой конденсации аминокислот наиболее всего подходят активированные эфиры -защищенных аминокислот, например, -гидроксисукцинимидный эфир или 2,4,5-трихлорфенильный эфир. Для эффективного катализа аминолиза используют гидроксисоединения, обладающие кислотностью приблизительно равной кислотности уксусной кислоты, например 1-гидроксибензотриазол. В качестве промежуточных аминозащитных групп предложены группы, удаляемые гидрированием, например бензилоксикарбонильный остаток ( -остаток) или отщепляемые в слабой кислоте группы. В качестве защитных групп для -аминогруппы используют,например, следующие группы трет-бутилоксикарбонил, флуоренилметилоксикарбонил,карбобензокси, соответственно карбобензтио (по выбору с п-бром- или п-нитробензильным остатком), трифторацетил, фталильный остаток, о-нитрофеноксиацетил, тритил, п-толуолсульфонил, бензил, бензил, имеющий заместители в бензольном кольце (п-бром- или пнитробензильный остаток) и а-фенилэтил. Кроме того, методы защиты аминогрупп описаны в следующих книгах. ,.(Химия аминокислот),, 1961,, . - Т.2, например, стр. 883 и др.,(Принципы пептидного синтеза),, 1984.(Твердофазный синтез пептидов).,., , , 1984..,(Пептиды). Гл.1. - С. 1-285, 1979.., а также(Пептиды). - Т. 2. / Под. ред. Е. , ,,. В основном эти же защитные группы используют и для защиты других функциональных боковых групп (ОН-групп,2-групп) соответствующих аминокислот. Имеющиеся гидроксигруппы (серина и треонина) предпочтительно защищают с помощью бензильных групп и аналогичных групп. Прочие не -аминогруппы (например,аминогруппы в -положении, гуанидиногруппы аргинина) предпочтительно защищают ортогонально. Отдельные производные аминокислот, за исключением лизина и орнитина, модифицированных 1 группами, имеются в продаже. Возможная схема процесса получения последних соединений является следующей 1. Амидируют -карбоксильную группу. 2. Защищают -аминогруппу -группой. 3. Защищают -аминогруппу Вос-группой, таким образом обеспечивая селективность последующего отщепления аминозащитных групп. 4. Отщепляют -группу с е-аминогруппы. 5. Присоединяют требуемую 1 группу по -аминогруппе. 6. Отщепляют Вос-группу от -аминогруппы. 7. Защищают -аминогруппу -группой. Для введения 1 группы с целью модификации аминогруппы лизина соответствующей карбоновой кислотой в основном используют способы, описанные для конденсации аминокислот. Однако наиболее предпочтительна конденсация с использованием карбодиимида, например 1-этил-3-(3-диметиламинопропил)карбодиимида, и 1-гидроксибензотриазола. Реакцию конденсации аминокислот проводят в обычных нейтральных растворителях и суспендирующих средствах (например в дихлорметане), причем по выбору для повышения растворимости может быть добавлен диметилформамид. В качестве синтетических материалов-носителей используют нерастворимые полимеры, например набухающую в органическом растворителе полистирольную смолу в форме гранул (например сополимеризат полистирола и 1 дивинилбензола). Синтез защищенного декапептидамида, имеющего после отщепления от носителя в присутствиитребуемую С-концевую амидную группу, на метилбензгидриламидной смоле (МВНА-смола,т.е. полистирольная смола, содержащая метилбензгидриламидные группы) может быть осуществлен согласно по постадийной схеме (таблица 1). 7 защищенные аминокислоты обычно конденсируют в присутствии трехкратного молярного избытка диизопропилкарбодиимидаи 1-гидроксибензотриазола в СН 2 С 2/ДМФ в течение 90 мин, а Вос-защитную группу отщепляют под действием 50 -ной трифторуксусной кислоты (ТФУ) в 22. Для контроля полноты прохождения реакции используют хлоранильный тест по Кристенсену и нингидриновый тест по Кайзеру. Остаток свободной аминогруппы блокируют ацетилированием в присутствии пятикратного избытка ацетилимидазола в СН 2 С 2. Последовательность реакционных стадий наращивания пептидной цепи на смоле приведена на постадийной схеме. Для отщепления связанного со смолой пептида конечный продукт твердофазного синтеза сушат в вакууме над Р 2 О 5 и обрабатывают 500-кратным избытком смеси /анизол 101/об.об. в течение 60 мин при 0 С. После упариванияи анизола в вакууме пептидамид выпадает в осадок при перемешивании с безводным этиловым эфиром в виде твердого вещества белого цвета, отделение от осадка полимерного носителя осуществляют промыванием 50 -ной водной уксусной кислотой. После осторожного концентрирования раствора в уксусной кислоте в вакууме пептид может быть получен в виде очень вязкого масла, которое при добавлении абсолютного эфира на холоде превращается в твердое вещество белого цвета. Дальнейшую очистку проводят известным методом высокоэффективной жидкостной хроматографии (ВЭЖХ). Перевод пептида в его кислотно-аддитивную соль может быть осуществлен взаимодействием с кислотами по известному способу. И наоборот, свободный пептид может быть получен взаимодействием его кислотно-аддитивной соли с основанием. Эмбонат пептида может быть получен взаимодействием соли пептида и трифторуксусной кислоты(соль с ТФУ) с эмбоновой кислотой (памовой кислотой) или соответствующей динатриевой солью эмбоновой кислоты. Для этого соль пептида с ТФУ в водном растворе обрабатывают раствором динатриевой соли эмбоновой кислоты в полярном апротонном растворителе, предпочтительно диметилацетамиде и выделяют образующийся осадок светло-желтого цвета. Следующие примеры приведены для пояснения изобретения без ограничения области изобретения. Пример 1.(2)12(3)3-456-7-8-910-2 Синтез проводят согласно постадийной схеме твердофазного синтеза (см. протокол пептидного синтеза, стр. 11) с использованием конденсации в присутствии /) и 3,3 г смолы МВНА (плотность нагрузки 1,08 ммоль/г). После отщепления от полимерного носителя в присутствииобразуется неочищенный пептид (3,4 г), который затем очищают стандартным методом препаративной ВЭЖХ. После лиофильного высушивания получают 1,43 г чистого по данным ВЭЖХ продукта брутто-формулы С 72, Н 96, 17, 14, с соответствующими масс-спектром, МС-ББА 1458,7 (МН) (рассчитано 1457,7) и 1 Н-ЯМР-спектром. 1 Н-ЯМР (500 МГц, 2/-6,часть/млн) 8,7-7,2 (несколько , ароматический Н инеполного обмена), 6,92 и 6,58 (2, 22, ароматический Н ), 5,2-3,5 (несколько , С-Н и алифатический Н), 3,2-2,6 (несколько , ароматический С-Н), 2,1-0,7(несколько , остаточный алифатический Н), 1,70 (, 3 Н, ацетил), 1,20 (, 3 Н, С-Н ),0,8 (, -, ). Ацетатную форму декапептида из примера 1 могут получать описанным выше способом или известными в литературе методами. Результаты ЯМР 1 Н-ЯМР (500 МГц, ДМСО-6,часть/млн) 8(2)12(3)3-456-7-8-9102 Синтез проводят согласно постадийной схеме твердофазного синтеза (см. протокол пептидного синтеза, стр. 11) с использованием конденсации в присутствии /) и 4,0 г смолы МВНА (плотность нагрузки 1,11 ммоль/г). После отщепления от полимерного носителя в присутствииобразуется неочищенный пептид (4,87 г), который затем очищают стандартным методом препаративной ВЭЖХ. После лиофильного высушивания получают 0,93 г чистого по данным ВЭЖХ продукта, который превращают в требуемое соединение в присутствии 4-амидинофениламино-4-оксомасляной кислоты и ВОР в качестве конденсирующего агента. После повторной очистки методом ВЭЖХ получают 148 мг конечного соединения брутто-формулы С 85, Н 112, 17, 15,с соответствующими масс-спектром, - 1647,6 (МН) (рассчитано 1645,8) и 1 Н-ЯМР-спектром. 1 Н-ЯМР (500 МГц, ДМСО-6,часть/млн) 10,4 (, 1) и 9,13 (, 2) и 8,94 (, 2,из 4-амидиноанилина), 8,6-7,35 (несколько , ароматические Н и ), 7,22 и 7,18 (2, 4,ароматический Н ), 6,95 и 6,58 (2, 4, ароматический Н ), 5,2-3,5 (несколько(2)12(3)3-456-7-8-910-2 Синтез проводят согласно постадийной схеме твердофазного синтеза (см. протокол пептидного синтеза, стр. 11) с использованием конденсации в присутствии /) и 4,0 г смолы МВНА (плотность нагрузки 0,97 ммоль/г). После отщепления от полимерного носителя в присутствииобразуется неочищенный пептид (4,0 г), который затем очищают стандартным методом препаративной ВЭЖХ. После лиофильного высушивания получают 1,39 г чистого по данным ВЭЖХ продукта, который превращают в требуемое соединение в присутствии 4-амидинофениламино-4-оксомасляной кислоты и ВОР в качестве конденсирующего агента. После повторной очистки методом ВЭЖХ получают 440 мг конечного соединения брутто-формулы С 82, Н 106, 19, 15,с соответствующими масс-спектром, - 1632,7 (МН) (рассчитано 1631,7) и 1 Н-ЯМР-спектром. 1 Н-ЯМР (500 МГц, ДМСО-6,часть/млн) 10,4 (, 1) и 9,15 (, 2) и 9,0 (, 2,из 4-амидиноанилина), 8,60 (, 2, ароматический Н), 8,3-7,2 (несколько , ароматические Н и ), 7,27 и 7,20 (2, 4 Н, ароматический Н ), 6,96 и 6,60 (2, 4 Н, ароматический Н ), 5,2-3,5 (несколько , С-Н и алифатический Н), 3,2-2,4 (несколько ,- и -СН 3), 2,1-1,1 (несколько , остаточный алифатический Н), 1,70 (, 3 Н, ацетил),1,20 (, 3 Н, С-Н ), 0,85 (, 6, С-Н ). Пример 4.(2)12(3)3-456-7-8-910-2 Синтез проводят согласно постадийной схеме твердофазного синтеза (см. протокол пептидного синтеза, стр. 11) с использованием конденсации в присутствии /) и 2,5 г смолы МВНА (плотность нагрузки 1,08 ммоль/г). После отщепления от полимерного 9 7934 1 2006.04.30 носителя в присутствииобразуется неочищенный пептид (2,78 г), который затем очищают стандартным методом препаративной ВЭЖХ. После лиофильного высушивания получают 400 мг чистого по данным ВЭЖХ продукта брутто-формулы С 75, Н 102, 15, 14, с соответствующими масс-спектром, МС- 1472,6 (рассчитано 1471,7) и 1 Н-ЯМР-спектром. 1 Н-ЯМР (500 МГц, 2/Д-6,часть/млн) 8,62 (, 2), 8,30 (, 2 Н), 7,80 (,4 Н), 7,66 (, 1 Н), 7,47 (, 2 Н), 7,36 (, 1, ароматический Н), 7,25 и 7,20 (2, 4, ароматический Н ), 6,96 и 6,63 (2, 4, ароматический Н ), 5,10-4,0 (несколько , СН и алифатический Н), 3,75-2,65 (несколько , - и -СН 3), 2,1-1,05 (несколько , остаточный алифатический Н), 1,74 (, 3 Н, ацетил), 1,23 (, 3 Н, - ), 1,20 (, 3 изопропил), 0,8 (, 3 Н, С-Н ). Пример 5.(2)12(3)3-456-7-8-9-10-2 Синтез проводят согласно постадийной схеме твердофазного синтеза (см. протокол пептидного синтеза, стр. 11) с использованием конденсации в присутствии /) и 2,5 г смолы МВНА (плотность нагрузки 1,08 ммоль/г). После отщепления от полимерного носителя в присутствииобразуется неочищенный пептид (2,74 г), который затем очищают стандартным методом препаративной ВЭЖХ. После лиофильного высушивания получают 840 мг чистого по данным ВЭЖХ продукта брутто-формулы С 75, Н 102, 15, 14, с соответствующими масс-спектром, МС- 1472,6 (МН) (рассчитано 1471,7) и 1 Н-ЯМР-спектром. 1 Н-ЯМР (500 МГц, 2/ДМСО-6,часть/млн) 8,6 (, 2 Н), 8,3 (, 2 Н), 7,85 (,2 Н),7,8 (, 2 Н), 7,65 (, 1 Н), 7,46 (, 2 Н), 7,35 (, 1, ароматический Н), 7,23 и 7,17 (2,4, ароматический Н ), 7,0 и 6,6 (2, 4, ароматический Н ), 5,10-3,8 (несколько , С-Н и алифатический Н), 3,75-2,6 (несколько , - и -3), 2,1-1,05 (несколько , остаточный алифатический Н), 1,70 (, 3 Н, ацетил), 1,23 (, 3 Н, - ), 1,20 (,3 изопропил), 0,8 (, 3 Н, С-Н ). Пример 6. 3-(4-фторфенил)пропионила(1)1-4 Ме-56-7-8-9-10-2 Синтез проводят согласно постадийной схеме твердофазного синтеза (см. протокол пептидного синтеза, стр. 11) с использованием конденсации в присутствии /) и 9,2 г смолы МВНА (плотность нагрузки 1,08 ммоль/г). После отщепления от полимерного носителя в присутствииобразуется неочищенный пептид (5,8 г), который затем очищают стандартным методом препаративной ВЭЖХ. После лиофильного высушивания получают 2,0 г чистого по данным ВЭЖХ незамещенного октапептида, 0,4 ммоль которого превращают в неочищенный требуемый продукт (790 мг) в присутствии 0,5 ммоль 3 амино-1,2,4-триазол-5-карбоновой кислоты и РуВОР в качестве конденсирующего агента. После повторной очистки методом ВЭЖХ получают 200 мг конечного продукта бруттоформулы С 64, Н 86, 17, 12,соответствующим масс-спектром, МС-ББА 1304,6(, 2 Н), 4,41 (, 1 Н), 4,30-4,05 (несколько , 4 Н, С-Н), 3,66-2,25 (несколько , алифатические и ароматические Н в боковой цепи), 2,95 и 2,75 (, -), 2,05-1,1 (несколько ,остаточный алифатический Н), 1,20 (, - ), 0,75 (, 6, С- ). Соединения по настоящему изобретению формулыисследуют по их связыванию с рецептором. Использованные способы относятся к известным методам, описанным в ста 10 7934 1 2006.04.30 тье., , (1995) т. 231, стр. 535-543. Цетрореликс, синтезированный, как описано выше, иодируют 125 (производства фирмы , удельная активность 80,5 Бк/фмоль) с использованием реагента(производства фирмы ). Реакционную смесь очищают методом обращеннофазовой высокоэффективной жидкостной хроматографии, при этом получают моноиодированный цетрореликс без примеси немеченного пептида. При необходимости приблизительно 80125-цетрореликса и немеченного соединения по изобретению являются пригодными для специфического связывания с рецептором. Соединения по настоящему изобретению могут быть исследованы по их действиюс использованием следующих методов 1 и 2, причем аффинность связывания определяют методом анализа связывания с 125-цетрореликсом (метод 1), а функциональную активность определяют с использованием трипторелина в качестве агонистического стимулятора (метод 2). Метод 1. Анализ связывания с рецептором по статьеТ.,К.,.,.(Отбор и характеристика линий клеток млекопитающих, обладающих устойчивой суперэкспрессией рецепторов гонадолиберина гипофиза человека) (1995), т. 231, стр. 535-543. Для исследования связывания с рецептором цетрореликс иодируют 125 (производства фирмы , удельная активность 80,5 Бк/фмоль) с использованием реагента(производства фирмы ). Реакционную смесь очищают методом высокоэффективной жидкостной хроматографии с замещенными фазами, причем получают моноиодированный цетрореликс без примеси немеченного пептида. Приблизительно 80125-цетрореликса являются пригодными для специфического связывания с рецептором. Анализ связывания с рецептором проводят с использованием интактных клеток в физиологических условиях по описанной методике (. 1995). Субконфлюентные культуры стабильных трансфектных клеток , которые экспрессируют на поверхности рецепторчеловека, отделяют инкубированием в присутствии / (137 мМ, 2,7 мМ , 8,1 мМ 24, 11,47 мМ КН 2 РО 4)/1 мМ ЭДТУ и собирают центрифугированием. Осадок клеток ресуспендируют в буфере для связывания ( без Н 2 СО 3, содержащая 4,5 г/л глюкозы, 10 мМрН 7,5, 0,5 мас./об.БСА, 1 г/л бацитрацина, 0,1 г/л , 0,1 мас./об.3. Для анализа по механизму замещения суспензию 0,25106 клеток/100 мкл, содержащую приблизительно 225 пМ 125-цетрореликс(удельная активность 5-10105 импульс/мин/пмоль), инкубируют в присутствии различных концентраций немеченного соединения по настоящему изобретению в качестве конкурирующего соединения. Суспензию клеток в 100 мкл среды для связывания наслаивают в трубки для исследования объемом 400 мкл на 200 мкл смеси 84 об.силиконового масла ( Тур 550)/16 об.парафинового масла. После инкубирования в течение 1 ч при 37 С при непрерывном медленном встряхивании клетки отделяют от инкубационной среды путем центрифугирования в течение 2 мин при 9000 об/мин (центрифуга 13,8, производства, , Германия). Верхние части трубок, содержащие осадок клеток, отрезают. Затем в осадке клеток и надосадочной жидкости определяют число импульсов -излучения. Количество неспецифически связанных соединений, включая немеченный цетрореликс при конечной концентрации 1 мкМ, которое в типичном случае составляет 10 от общего количества связанных соединений. Для анализа данных по связыванию используют аналитическую программу / ( 3,0). Метод 2. Функциональный анализ для определения антагонистического действия. Анализ проводят с некоторыми модификациями, как описано в статьеТ., .,.-11 7934 1 2006.04.30(Характеристика аналогов релизинг-гормона гонадотропина, основанная на чувствительном анализе репортерного гена клеточной люциферазы) (1997), , т. 251, стр. 17-23 (. 1997). 10000 клеток в лунке, которые содержат на поверхности -рецептор человека и репортерный ген люциферазы человека, культивируют в микропланшете в течение 24 ч с использованием средыс добавками и 1 сыворотки теленка . Затем клетки стимулируют в течение 6 ч 1 нМ раствором -6 . Перед стимулированием добавляют антагонистические соединения по настоящему изобретению и наконец для количественного определения активности клеточной люциферазы клетки лизируют. Рассчет величины 50 проводят с использованием кривой дозо-зависимого действия и нелинейного регрессионного анализа - (программа 2,0, . , , ). Количественное определение активности люцтферазы в основном определяют по известному методу (,101/161) с использованием дубликатов и современных систем для анализа люциферазы ( 4030). При добавлении коэнзима А (СоА) происходит окисление люциферил-СоА с благоприятной кинетикой. После удаления культуральной среды из микропланшета клетки лизируют путем добавления 100 мкл буфера для лизиса (25 мМ трис-фосфат, рН 7,8, 2 мМ дитиотреитол, 2 мМ 1,2 диаминоциклогексан--тетрауксусная кислота , 10(об./об.) глицерин,1(об./об.) тритон Х-100). После инкубирования в течение 15 мин при комнатной температуре 10 мкл клеточного лизата переносят в микропланшет белого цвета, пригодные для люминометрического определения . Ферментативную реакцию инициируют путем добавления 50 мкл буфера для анализа (20 мМ трицин, рН 7,8, 1,07 мМ(3)42, 2,67 мМ 4, 0,1 мМ этилендиаминтетрауксусная кислота (ЭДТУ),33,3 мМ дитиотреитол, 270 мкМ коэнзим А, 470 мкМ люциферин из светляков, 530 мкМ 2). Через 1 мин определяют люминисценцию в течение общего времени 1 сек и с временем сброса сигнала 5 мин при использовании 96 Р, . Таким образом получают данные действия, представленные в таблице 2, причем аффинность связывания представлена величинами , функциональная активность величинами 50, а термин пМ означает пикомоль в литре. Таблица 1 Постадийная схема (протокол пептидного синтеза) Стадия 1 2 3 4 5 6 7 8 9 Функция Промывка Промывка Отщепление Промывка Промывка Промывка Нейтрализация Промывка Промывка Растворитель/реагент об./об. Метанол ДХМ ДХМ/ТФУ (11) Изопропанол Метанол ДХМ ДХМ/ (91) Метанол ДХМ Добавление Восаминокислоты в ДХМДХМ, по выбору ДХМ/ Метанол ДХМ 7934 1 2006.04.30 Таблица 2 Соединение Цетрореликс Пример 1 (ацетат) Пример 2 Пример 3 Пример 6 число независимых друг от друга испытаний. Национальный центр интеллектуальной собственности. 220034, г. Минск, ул. Козлова, 20. 13
МПК / Метки
МПК: C07K 7/23, A61P 35/00, A61K 38/09
Метки: улучшенной, растворимостью, антагонисты
Код ссылки
<a href="https://by.patents.su/13-7934-antagonisty-lhrh-s-uluchshennojj-rastvorimostyu.html" rel="bookmark" title="База патентов Беларуси">Антагонисты LHRH с улучшенной растворимостью</a>
Новые антагонисты ЛГ-РФ-рецепторов с повышенной эффективностью
Номер патента: 5834
Опубликовано: 30.12.2003
Авторы: ЭМИГ Петер-Пауль, КЛЕННЕР Томас, КУЧЕР Бернхард, БЕКЕРС Томас, КАРПЕНТИР Патрисия-Мари, БЕРНД Майкл
МПК: A61K 38/08, A61K 38/04, A61P 35/00...
Метки: повышенной, новые, эффективностью, лг-рф-рецепторов, антагонисты
Текст:
...з-аминогруппе В-лизина или В-орнитина в положении 6 и конденсация полученного соединения с карбоновой кислотой общей формулы Х/Пк 4-соон (х/п) где К 4 имеет указанные в п. 1 значения, 3) отщепление защитной группы по оъ-аминогруппе В-лизина или В-орнитина, Ь) повторение стадий с) и 01) с аминокислотами 1-5 общей формулы У, в последовательности 5-1,1) отщепление соединения, полученного на стадии 11), от твердого носителя и...
Производные хинолина как антагонисты NK3 рецептора тахикинина

Номер патента: 5329
Опубликовано: 30.09.2003
Авторы: ФАРИНА, Карло, РАВЕЛИЯ, Лука, Франческо, ДЖАРДИНА, Джузеппе, Амальдо, Мария, ГРУНЬИ, Марио
МПК: A61P 13/00, C07D 215/16, A61K 31/47...
Метки: производные, тахикинина, рецептора, хинолина, антагонисты
Пептидные антагонисты бомбезина их фармацевтически приемлемые кислоты или соли , фармацевтическая композиция, обладающая антагонистической активностью по отношению к бомбезину, и способ лечения рака у млекопитающих

Номер патента: 3837
Опубликовано: 30.03.2001
Авторы: КЭЙ, Рен Жи, СКЭЛЛИ, Эндрю В.
МПК: C07K 7/06, A61K 37/02, A61K 38/08...
Метки: фармацевтическая, приемлемые, бомбезина, фармацевтически, способ, пептидные, обладающая, соли, антагонисты, активностью, отношению, млекопитающих, кислоты, композиция, лечения, бомбезину, антагонистической, рака, или
Текст:
...или 1-С 10-алкилом А 1 является -, -, - или -, -, - или -, или 2 являетсяилииявляется 2. 3. Пептидный антагонист бомбезина по п. 2, где А 9 является Тас. 4. Пептидный антагонист бомбезина по п. 2, где А 9 является . 5. Пептидный антагонист бомбезина по п. 3, где Х является Н или Ас, А 1 является -, - или -, а 2 А является . 6. Пептидный антагонист бомбезина по п. 4, где Х является Н или Ас, А 1 является , - или -, а А 2 является . 7....
Металлокорд с улучшенной фиксацией проволок сердечника и устройство для его изготовления

Номер патента: U 967
Опубликовано: 30.09.2003
Авторы: Савенок Анатолий Николаевич, Веденеев Александр Владимирович, Желтков Александр Сергеевич, Филиппов Вадим Владимирович
Метки: изготовления, металлокорд, улучшенной, сердечника, проволок, фиксацией, устройство
Текст:
...но в некоторых случаях - также цинкового или полимерного покрытия. Покрытие должно обладать хорошим сцеплением как с резиной, так и со стальной основой проволоки. Корд, согласно полезных моделей, может состоять из проволоки разных диаметров. Наиболее часто используемый диапазон диаметров проволок, из которых свивают корд для армирования резинотехнических изделий, составляет от 0,10 до 0,80 мм. Шаги свивки металлокорда при этом обычно выбирают...
Металлокорд с улучшенной фиксацией проволок сердечника и устройство для его изготовления

Номер патента: U 782
Опубликовано: 30.03.2003
Авторы: Савенок Анатолий Николаевич, Филиппов Вадим Владимирович, Веденеев Александр Владимирович, Желтков Александр Сергеевич
Метки: фиксацией, сердечника, проволок, металлокорд, улучшенной, изготовления, устройство
Текст:
...сердечника и полнота проникновение резины в структуру корда обеспечивается, если стальные проволоки имеют хорошее сцепление (адгезию) с резиной. Преимущественно это достигается при нанесении на проволоку латунного покрытия, но в некоторых случаях также цинкового или полимерного покрытия. Покрытие должно обладать хорошим сцеплением как с резиной, так и со стальной основой проволоки. Корд, согласно полезной модели, может состоять из...
Предыдущий патент: Рабочий орган землеройной машины
Следующий патент: Способы получения промежуточных продуктов, используемых при производстве пестицидов
Случайный патент: Способ лечения гипофункции яичников у женщин больных ревматоидным артритом