Инверсионно-вольтамперометрический способ определения алюминия
Номер патента: 16368
Опубликовано: 30.10.2012
Авторы: Кулак Анатолий Иосифович, Матвейко Николай Петрович, Ксензова Юлия Николаевна





Текст
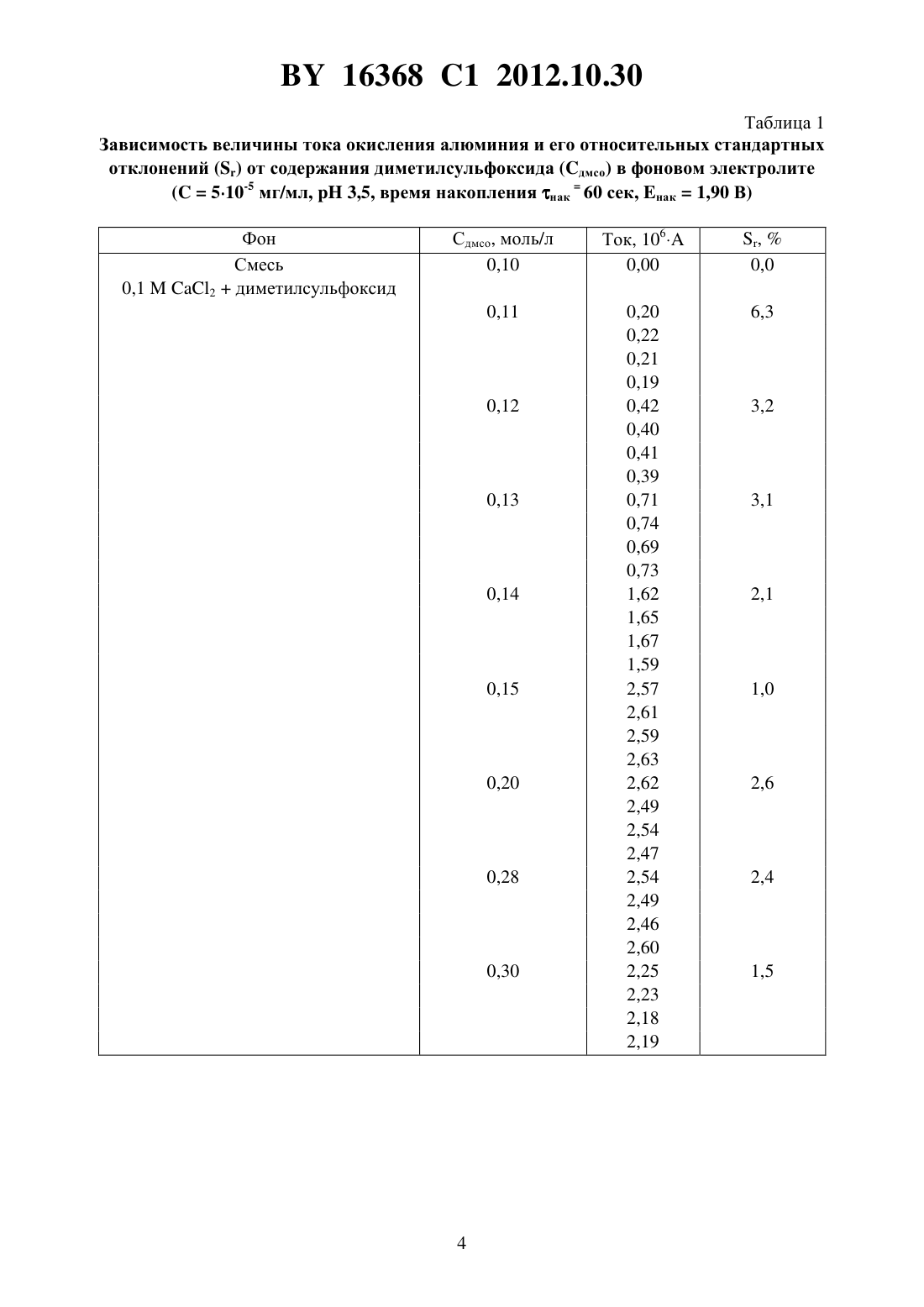
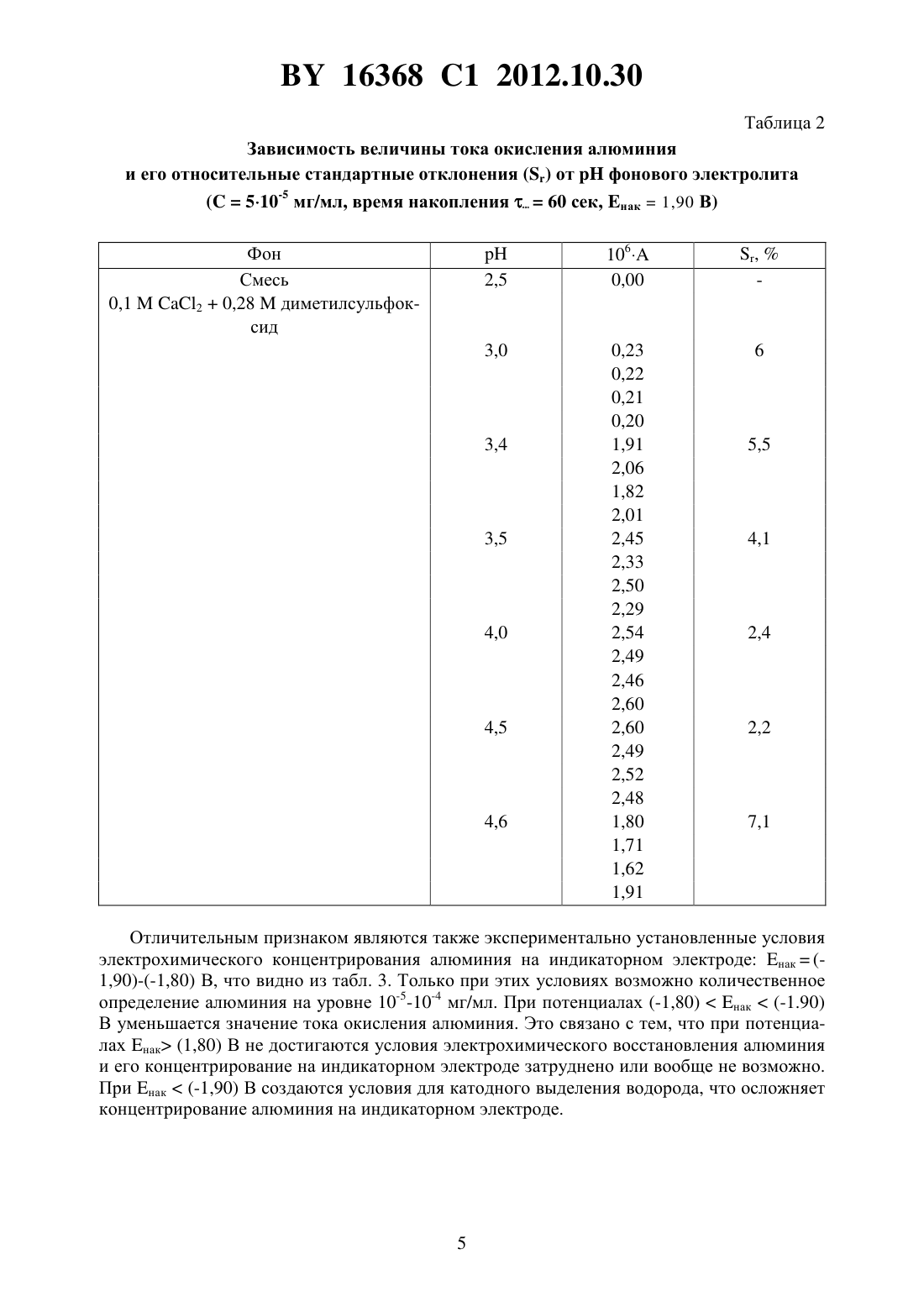
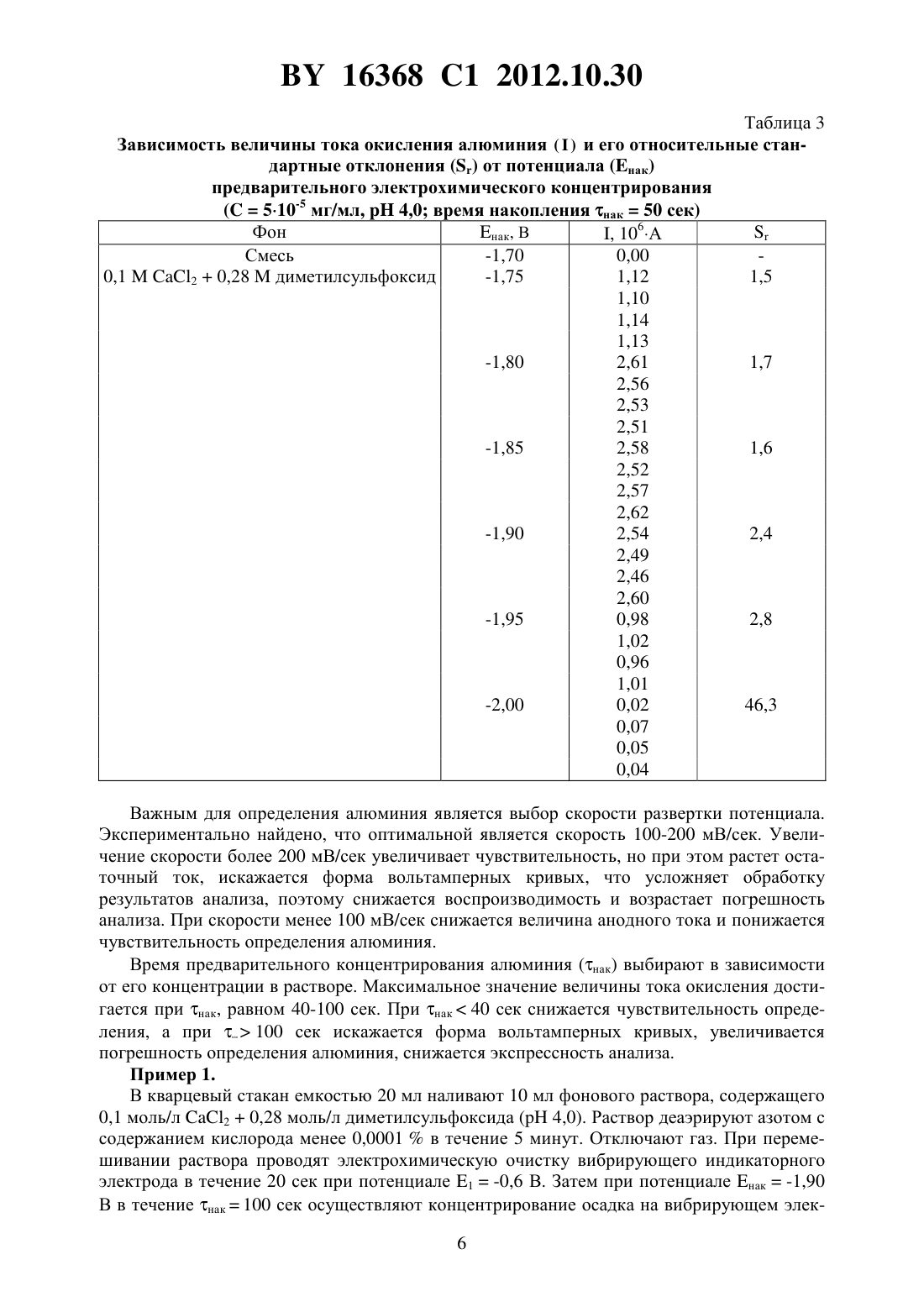
(51) МПК НАЦИОНАЛЬНЫЙ ЦЕНТР ИНТЕЛЛЕКТУАЛЬНОЙ СОБСТВЕННОСТИ(71) Заявитель Государственное научное учреждение Институт общей и неорганической химии Национальной академии наук Беларуси(72) Авторы Кулак Анатолий Иосифович Матвейко Николай Петрович Ксензова Юлия Николаевна(73) Патентообладатель Государственное научное учреждение Институт общей и неорганической химии Национальной академии наук Беларуси(57) Инверсионно-вольтамперометрический способ определения содержания алюминия в водном растворе, при котором осуществляют концентрирование осадка на поверхности ртутного пленочного электрода при потенциале от -1,9 до -1,8 В в течение 40-100 сек в фоновом электролите и исследуемом растворе, содержащих 0,1-0,2 моль/л 2, 0,15-0,28 моль/л диметилсульфоксида и хлороводородную кислоту до 3,5-4,5, с последующей регистрацией вольтамперных кривых фонового электролита и исследуемого раствора при скорости развертки потенциала 100-200 мВ/с и определяют концентрацию алюминия в исследуемом растворе по разности высот анодных пиков в интервале потенциала от -1,2 до -1,0 В относительно насыщенного хлорсеребряного электрода сравнения. Изобретение относится к области аналитической химии, в частности к инверсионновольтамперометрическому (ИВА) способу определения алюминия, и может найти применение для определения алюминия в пищевой продукции, объектах окружающей среды,биологических жидкостях, технологических средах. Известны ИВА-способы определения алюминия, основанные на связывании ионов алюминияв комплекс с органическими лигандами аммонийной солью нитрозофенилгидроксиламина - купфероном 1, 2, солохромом фиолетовым 3, ализарином( ) 4, пирогаллоловым красным () 5, 1,2-дигидроксиантрахинон-3-сульфоновой кислотой (1,2-3- ) 6, 7. В качестве индикаторного электрода использовался ртутнокапельный 2-6 либо пленочный ртутный 7 или висмутовый 1 электрод. Все эти способы относятся к косвенным, поскольку ИВА-анализу подвергаются не ионы, а металлокомплексные алюминийорганические соединения, адсорбированные на поверхность зондового (рабочего) электрода. В этих способах определение количества адсорбированного алюминиевого металлокомплекса осуществляется путем регистрации пика 16368 1 2012.10.30 катодного тока на поляризационных потенциодинамических кривых либо методом дифференциально-импульсной, либо квадратно-волновой вольтамперометрии 8. Одним из существенных недостатков этих способов является то, что на стадии регистрации пика катодного тока мешающее действие могут оказывать любые компоненты анализируемого раствора, способные восстанавливаться при потенциалах вблизи потенциала восстановления алюминиевого металлокомплекса 9, а именно растворенный кислород, ряд катионов металлов. Для указанных способов характерны и другие существенные недостатки, а именно,необходимость введения органических индикаторных соединений (купферон, солохром,ализарин и др.) в анализируемый раствор, наличие дополнительных стадий образования комплексов данных соединений с ионами алюминия и их адсорбции на поверхность электрода, понижение селективности определений ввиду возможного химического взаимодействия мешающих ионов с индикаторными соединениями. Введение индикаторных соединений, как правило, ароматических органических веществ, существенно повышает токсичность анализируемых растворов. Необходимость образования комплексных химических соединений алюминия с индикаторными красителями часто вносит неопределенность в результаты анализа, поскольку, как правило, алюминий с этими лигандами образует соединения переменного состава 10. Наличие адсорбционной стадии также является источником неопределенностей и, соответственно, погрешностей, поскольку при наличии других (мешающих) компонентов в анализируемом растворе они могут конкурировать с комплексными ионами алюминия за адсорбционные места на поверхности электрода 1. Результатом этого является сильная нелинейная зависимость аналитического сигнала (катодного тока) от концентрации вводимого индикаторного соединения (в частности, купферона, ализарина либо пирогаллолового красного). Использование ртутного капельного электрода 2-6 является дополнительным существенным недостатком в связи с необходимостью извлечения капель ртути из анализируемого раствора по завершению процесса анализа. Наиболее близким к предлагаемому изобретению является ИВА-анализ алюминия в присутствии 1,2-дигидроксиантрахинон-3-сульфоновой кислоты с использованием ртутного пленочного электрода 7 (прототип). В этом методе детектирование алюминия осуществляется по пику тока при -1,15 В (относительно насыщенного хлорсеребряного электрода сравнения) порог чувствительности 1 мкмоль/л алюминия и рабочая (линейная) область концентраций алюминия от 1 до 20 мкмоль/л, что соответствует по порогу чувствительности 2,710-5 мг/мл и по области линейности от 2,710-5 до 5,410-4 мг/мл в пересчете на алюминий. Однако этот метод требует обязательного введения в анализируемый раствор добавки 1,2-дигидроксиантрахинон-3-сульфоновой кислоты (лиганда), и, кроме того, включает обязательную стадию образования комплексного соединения между алюминием и упомянутым лигандом и обязательную стадию адсорбции этого комплексного соединения на поверхность ртутного пленочного электрода. Задачей изобретения является повышение чувствительности, точности определения и воспроизводимости результатов способа ИВА-анализа алюминия в водном растворе. Поставленная задача достигается заявляемым инверсионно-вольтамперометрическим способом определения содержания алюминия в водном растворе путем электрохимического концентрирования непосредственно алюминия на поверхности ртутного пленочного электрода при потенциалах (-1,90)-(-1,80) В в течение 40-100 сек в растворе (0,1-0,2)2(0,15-0,28)диметилсульфоксида в присутствии хлороводородной кислотыс 3,5-4,5 с последующей регистрацией анодных пиков анализируемого раствора и фонового электролита при скорости развертки потенциала 100-200 мВ/с, расчетом содержания алюминия в растворе по разности высоты анодных пиков на вольтамперных кривых в 16368 1 2012.10.30 интервале потенциалов (-1,2)-(-1,0) В относительно насыщенного хлорсеребряного электрода сравнения. В предлагаемом способе прямого ИВА-анализа алюминий в результате катодной поляризации электрохимически концентрируется на поверхности ртутного пленочного электрода с последующей регистрацией пиков анодного тока на поляризационных потенциодинамических кривых. Новым в этом способе является то, что прямое электрохимическое накопление алюминия ведут на ртутном пленочном электроде при потенциалах (-1,90)-(-1,80) В в течение нак 40-100 сек в растворе (0,1-0,2)2(0,15-0,28)диметилсульфоксида при 3,5-4,5, поддерживаемом с помощью добавки соляной (хлороводородной) кислоты регистрацию анодных вольтамперных кривых ведут при скорости развертки потенциала 100-200 мВ/с, а концентрацию алюминия определяют по высоте пика в интервале потенциалов (-1,2)-(-1,0) В относительно хлорсеребряного электрода. Зависимость аналитического сигнала от концентрации алюминия в предлагаемых условиях прямо пропорциональна диапазону определяемых концентраций от 10-5 до 10-4 мг/мл. Абсолютной новизной являются экспериментально подобранный фон (0,1-0,2)2(0,15-0,28)диметилсульфоксида, установленныйраствора, от чего зависит количественное определение алюминия. Выбор содержания 2 в фоновом электролите(0,1-0,2) моль/л обусловлен следующим. При концентрации менее 0,1 моль/л электрическая проводимость фонового электролита становится недостаточной для регистрации воспроизводимых вольтамперных кривых и регистрации аналитического сигнала,достаточного для определения алюминия с хорошей воспроизводимостью результатов. Увеличение содержания 2 в фоновом электролите более 0,2 моль/л приводит к искажению формы вольтамперной кривой, что затрудняет идентификацию аналитического сигнала окисления алюминия и делает невозможным проведение анализа на этих фонах. Заявляемый способ функционирует в отсутствие индикаторных добавок и не требует наличия стадии адсорбции комплексных соединений алюминия на поверхность индикаторного электрода, что ведет к упрощению анализа, снижению токсичности анализируемых растворов, экономии дорогостоящих индикаторных реагентов и повышению экспрессности способа ИВА определения алюминия. Из табл. 1 видно, что при содержании в фоновом электролите диметилсульфоксида менее 0,15 моль/л ток окисления алюминия уменьшается и практически исчезает, регистрация воспроизводимого аналитического сигнала становится невозможной, что связано с одновременным выделением водорода и алюминия в процессе его электрохимического концентрирования на индикаторном электроде. Содержание диметилсульфоксида в фоновом электролите более 0,28 моль/л нецелесообразно, т.к. не приводит к увеличению аналитического сигнала, повышению точности и экспрессности определения алюминия. Оптимальный диапазон 3,5-4,5 определяется фиксированием одного пика растворения алюминия (отсутствием дополнительных мешающих пиков) и высокой воспроизводимостью результатов. Из табл. 2 видно, что приниже 3,5 пик тока окисления алюминия практически отсутствует. Это связано с невозможностью концентрирования алюминия на электроде при таких условиях из-за интенсивного выделения водорода. Повышениеболее 4,5 приводит к уменьшению высоты пика, обусловленного окислением алюминия, что не позволяет проводить количественное определение алюминия. Причина этого состоит в том, что приболее 4,5 наблюдается усиление процесса гидролиза солей алюминия, в результате чего образуются основные соли алюминия, из которых затруднено его электрохимическое осаждение на поверхности электрода. 16368 1 2012.10.30 Таблица 1 Зависимость величины тока окисления алюминия и его относительных стандартных отклоненийот содержания диметилсульфоксида (дмсо) в фоновом электролите(510-5 мг/мл,3,5, время накопления нак 60 сек, нак 1,90 В) 16368 1 2012.10.30 Таблица 2 Зависимость величины тока окисления алюминия и его относительные стандартные отклоненияотфонового электролита Отличительным признаком являются также экспериментально установленные условия электрохимического концентрирования алюминия на индикаторном электроде нак(1,90)-(-1,80) В, что видно из табл. 3. Только при этих условиях возможно количественное определение алюминия на уровне 10-5-10-4 мг/мл. При потенциалах (-1,80)нак(-1.90) В уменьшается значение тока окисления алюминия. Это связано с тем, что при потенциалах нак (1,80) В не достигаются условия электрохимического восстановления алюминия и его концентрирование на индикаторном электроде затруднено или вообще не возможно. При нак(-1,90) В создаются условия для катодного выделения водорода, что осложняет концентрирование алюминия на индикаторном электроде. 16368 1 2012.10.30 Таблица 3 Зависимость величины тока окисления алюминия и его относительные стандартные отклоненияот потенциала (нак) предварительного электрохимического концентрирования(510-5 мг/мл,4,0 время накопления нак 50 сек)-2,00 0,02 46,3 0,07 0,05 0,04 Важным для определения алюминия является выбор скорости развертки потенциала. Экспериментально найдено, что оптимальной является скорость 100-200 мВ/сек. Увеличение скорости более 200 мВ/сек увеличивает чувствительность, но при этом растет остаточный ток, искажается форма вольтамперных кривых, что усложняет обработку результатов анализа, поэтому снижается воспроизводимость и возрастает погрешность анализа. При скорости менее 100 мВ/сек снижается величина анодного тока и понижается чувствительность определения алюминия. Время предварительного концентрирования алюминия (нак) выбирают в зависимости от его концентрации в растворе. Максимальное значение величины тока окисления достигается при нак, равном 40-100 сек. При нак 40 сек снижается чувствительность определения, а при 100 сек искажается форма вольтамперных кривых, увеличивается погрешность определения алюминия, снижается экспрессность анализа. Пример 1. В кварцевый стакан емкостью 20 мл наливают 10 мл фонового раствора, содержащего 0,1 моль/л 20,28 моль/л диметилсульфоксида ( 4,0). Раствор деаэрируют азотом с содержанием кислорода менее 0,0001 в течение 5 минут. Отключают газ. При перемешивании раствора проводят электрохимическую очистку вибрирующего индикаторного электрода в течение 20 сек при потенциале 1-0,6 В. Затем при потенциале нак-1,90 В в течение нак 100 сек осуществляют концентрирование осадка на вибрирующем элекнак 16368 1 2012.10.30 троде. Регистрацию анодной вольтамперной кривой проводят в интервале потенциалов от-1,5 В до -0,8 В при скорости развертки потенциала 200 мВ/сек. Отсутствие пиков свидетельствует о чистоте фона. В стакан с фоновым электролитом добавляют 0,02 мл стандартного раствора алюминия 510-3 мг/мл. При перемешивании раствора проводят электрохимическую очистку вибрирующего индикаторного электрода в течение 20 сек при потенциале 1-0,6 В. После этого проводят электрохимическое концентрирование осадка при потенциале нак-1,90 В в течение нак 100 сек на вибрирующем индикаторном электроде. Регистрацию анодной вольтамперной кривой осуществляют в интервале потенциалов от -1,5 В до -0,8 В при скорости развертки потенциала 200 мВ/сек. Пик анодного растворения алюминия регистрируется в диапазоне потенциалов от -1,2 до -1,0 В. В раствор вводят добавку 0,02 мл стандартного раствора алюминия концентрацией 510-3 мг/мл. При перемешивании раствора проводят электрохимическую очистку вибрирующего индикаторного электрода в течение 20 сек при потенциале 1-0,6 В. После этого проводят электрохимическое концентрирование осадка при потенциале нак-1,90 В в течение нак 100 сек на вибрирующем индикаторном электроде. Регистрацию анодной вольтамперной кривой осуществляют в интервале потенциалов от -1,5 В до -0,8 В при скорости развертки потенциала 200 мВ/сек. Фиксируемый пик анодного растворения алюминия увеличивается приблизительно в два раза. По разности вольтамперных кривых рассчитывают содержание алюминия в растворе. Время одного анализа не превышает 15 минут. Пример 2. В кварцевый стакан емкостью 20 мл наливают 10 мл фонового раствора, содержащего 0,1 моль/л 20,28 моль/л диметилсульфоксида ( 4,0). Раствор деаэрируют азотом либо аргоном с содержанием кислорода менее 0,0001 в течение 5 минут. Отключают газ. При перемешивании раствора проводят электрохимическую очистку вибрирующего индикаторного электрода в течение 20 сек при потенциале 1-0,6 В. Затем при потенциале нак-1,90 В в течение нак 40 сек осуществляют концентрирование осадка на вибрирующем электроде. Регистрацию анодной вольтамперной кривой проводят в интервале потенциалов от -1,2 В до -0,8 В при скорости развертки потенциала 200 мВ/сек. Отсутствие пиков свидетельствует о чистоте фона. В стакан с фоновым электролитом добавляют 0,2 мл стандартного раствора алюминия 510-3 мг/мл. При перемешивании раствора проводят электрохимическую очистку вибрирующего индикаторного электрода в течение 20 сек при потенциале 1-0,6 В. После этого проводят электрохимическое концентрирование осадка при потенциале нак-1,90 В в течение нак 40 сек на вибрирующем индикаторном электроде. Регистрацию анодной вольтамперной кривой осуществляют в интервале потенциалов от -1,5 В до -0,8 В при скорости развертки потенциала 200 мВ/сек. Пик анодного растворения алюминия регистрируется в диапазоне потенциалов от -1,2 до-1,0 В. В раствор вводят добавку 0,2 мл стандартного раствора алюминия концентрацией 510-3 мг/мл. При перемешивании раствора проводят электрохимическую очистку вибрирующего индикаторного электрода в течение 20 сек при потенциале 1-0,6 В. После этого проводят электрохимическое концентрирование осадка при потенциале нак-1,90 В в течение нак 40 сек на вибрирующем индикаторном электроде. Регистрацию анодной вольтамперной кривой осуществляют в интервале потенциалов от -1,5 В до -0,8 В при скорости развертки потенциала 200 мВ/сек. Фиксируемый пик анодного растворения алюминия увеличивается приблизительно в два раза. По разности вольтамперных кривых рассчитывают содержание алюминия в растворе. Время одного анализа не превышает 15 минут. Правильность методики с использованием предлагаемого способа определения алюминия проверена методом введено-найдено. Результаты представлены в табл. 4. 16368 1 2012.10.30 Таблица 4 Результаты проверки правильности методики методом введено-найдено Найдено Введено Среднее количе- Относительное Доверительный интервал алюминия, алюминия , ство алюминия в стандартное от- (при доверительной веромг/мл растворе, мг/мл клонение, ,ятности 0,95) , мг/мл мг/мл-5 1,79 1010 09,9910-5 0,02810-5 09,8410-5 09,8310-5 Из табл. 4 следует, что при содержании алюминия в анализируемом растворе в концентрации 110-5 мг/мл метод по изобретению обеспечивает высокий уровень точности и воспроизводимости. По прототипу предел обнаружения алюминия составляет 2,710-5 мг/мл и требует обязательного введения индикаторного реагента. Таким образом, предлагаемый метод характеризуется наличием существенных преимуществ по сравнению с прототипом обладает более высокой чувствительностью, высокой точностью определения и воспроизводимостью результатов. Предлагаемый способ прост, экспрессен, не требует применения дефицитных реагентов и большого количества реактивов. Он может быть применен для контроля качества сырья пищевых продуктов и фармакопейных препаратов, а также для анализа объектов окружающей среды. Источники информации 1..,.,.-. , 2006. - . 68. - . 1013-1019. 2..,.,.,.., 2006. - . 97. - . 176-180. 3.,.,.,... , 2006. - . 18. - . 2257-2262. 8.2000-241388 9.., 1991. - . 250. - . 256-276. 10. Тихонов В.Н. Аналитическая химия алюминия. - М. Наука, 1971. - 266 с. Национальный центр интеллектуальной собственности. 220034, г. Минск, ул. Козлова, 20. 8
МПК / Метки
МПК: G01N 27/49
Метки: способ, инверсионно-вольтамперометрический, определения, алюминия
Код ссылки
<a href="https://by.patents.su/8-16368-inversionno-voltamperometricheskijj-sposob-opredeleniya-alyuminiya.html" rel="bookmark" title="База патентов Беларуси">Инверсионно-вольтамперометрический способ определения алюминия</a>
Способ определения натрия в плодоовощных продуктах путем прямой потенциометрии

Номер патента: 14071
Опубликовано: 28.02.2011
Авторы: Ломако Светлана Валерьевна, Егорова Зинаида Евгеньевна, Шачек Татьяна Михайловна
МПК: G01N 27/26
Метки: потенциометрии, определения, путем, способ, натрия, продуктах, прямой, плодоовощных
Текст:
...натрия в плодоовощных продуктах путем прямой потенциометрии с применением -ионоселективного мембранного электрода, включающем подготовку пробы, измерение в ней потенциала-ионоселективного мембранного электрода и определение концентрации натрия в исследуемой пробе методом добавок, подготовку пробы осуществляют путем ее разбавления 0,02 раствором уксусной кислоты, а в качестве добавки используют 1 раствор хлористого натрия,...
Способ получения геля фосфата алюминия в качестве субстанции антацидного лекарственного препарата

Номер патента: 2514
Опубликовано: 30.12.1998
Авторы: Дрожденюк Анатолий Павлович, Стрельченок Олег Анатольевич, Чащин Вадим Леонидович, Петров Петр Тимофеевич, Царенков Валерий Минович, Макаренко Михаил Васильевич, Свиридов Олег Васильевич
МПК: C01B 25/36, A61K 33/06
Метки: алюминия, антацидного, препарата, геля, лекарственного, качестве, способ, получения, фосфата, субстанции
Текст:
...(молярное соотношение алюминий/фосфор равно 1,3) и доводят медленным прибавлением 1 М раствора гидроокиси натрия рН реакционной смеси до величины 6,0. Величину рН контролируют с помощью рН - метра. Осадок промывают до отсутствия ионов хлора. При необходимости полученный гель центрифугируют, осадок высушивают при 80-100. Получают 11,8 г фосфата алюминия. Выход 97 . Анализ целевого продукта проводился аналогично примеру 1 содержание А 1 -17,5...
Способ определения калия в плодоовощных продуктах путем прямой потенциометрии

Номер патента: 14070
Опубликовано: 28.02.2011
Авторы: Шачек Татьяна Михайловна, Ломако Светлана Валерьевна, Егорова Зинаида Евгеньевна
МПК: G01N 27/26
Метки: калия, способ, продуктах, потенциометрии, прямой, путем, определения, плодоовощных
Текст:
...и вследствие этого снижается точность и правильность получаемых результатов. Задача изобретения - повышение точности определения калия в плодоовощных продуктах методом потенциометрии. Поставленная задача достигается тем, что в способе определения калия в плодоовощных продуктах путем прямой потенциометрии с применением -ионоселективного мембранного электрода, включающем подготовку пробы, измерение в ней потенциала ионоселективного...
Способ получения паяемого покрытия на тонких пленках алюминия

Номер патента: 4492
Опубликовано: 30.06.2002
Авторы: Сокол Виталий Александрович, Гринис Лариса Михайловна
МПК: H01L 21/288
Метки: паяемого, пленках, тонких, алюминия, покрытия, способ, получения
Текст:
...осаждения никеля вводят фторид натрия или калия и создают слабокислую среду с рН 3,5-5 введением аминоуксусной кислоты. Аминоуксусная кислота является также комплексообразующим агентом, связывающим ионы никеля в растворимые комплексы и регулирующим скорость контактного обмена алюминия на никель. Контактное осаждение никеля проводят в водном растворе, содержащем соль никеля в количестве 20-60 г/л, натрий фтористый или калий фтористый в...
Способ получения анодного оксида алюминия со сквозной пористостью

Номер патента: 9186
Опубликовано: 30.04.2007
Авторы: Григоришин Иван Леонтьевич, Войтик Ольга Леонидовна, Делендик Кирилл Иванович
МПК: C25D 11/04, C25D 11/24, C25D 11/02...
Метки: получения, оксида, способ, анодного, пористостью, сквозной, алюминия
Текст:
...травят дополнительно напыленный слой алюминия. Совокупность указанных признаков обеспечивает однородное вскрытие пор в барьерном слое с сохранением неизменности структуры, физических и др. свойств анодного оксида алюминия. Принцип, лежащий в основе изобретения, заключается в следующем. При достижении фронтом роста анодного оксида алюминия границы раздела алюминий-проводящее покрытие происходит утонение барьерного слоя анодного оксида...
Предыдущий патент: Стенд для приемочных испытаний редуктора
Следующий патент: Способ формирования изображения на металлизированной алюминием поверхности рулонного полимерного материала и травильный раствор для его осуществления
Случайный патент: Аксиально-поршневая гидромашина