Способ получения производных изохинолиния
Номер патента: 1329
Опубликовано: 16.09.1996
Авторы: Рой Арчибальд Сваринген, младший, Дэвид Артур Йевелл, Джон Джозеф Саварез, Хассан Али Эль-Сайяд







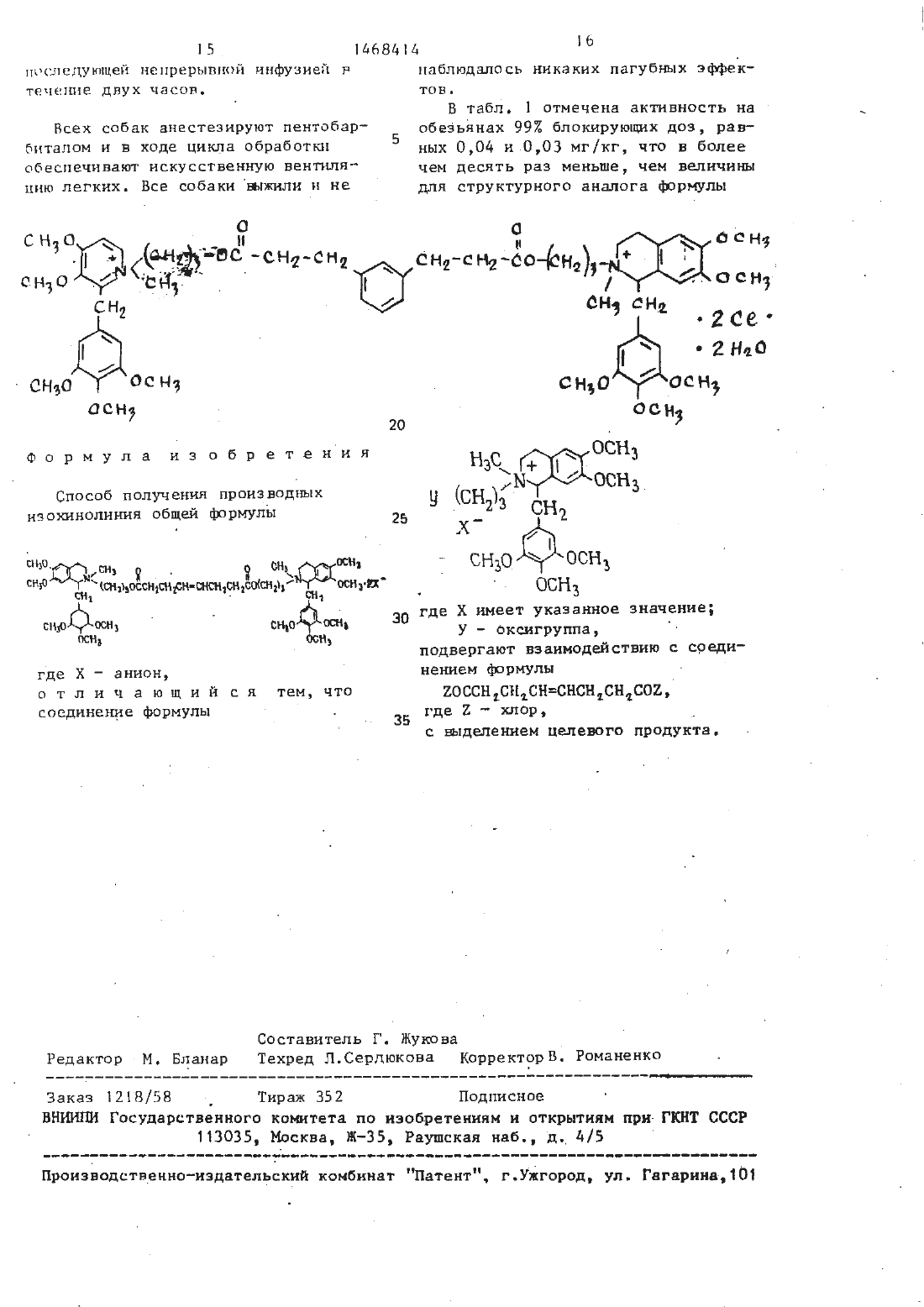
Текст
280 им. Хотя время удерживания (ВТ) меняется в завнсимостнот ряда факТОРОВ, ПОРЯДОК ЗЛЮНРОЕЗНИЯ ЯВЛЯЕТСЯ следукщнм, с(окситриметилен 5-бис- (1 К,25)ттриметоксибенэил)изонинолинииднхлорид 751 Все сложные диэфиры сушат до постоянной массы при давлении 0,1 мм ртст И температуре окружающей орет дм. Вращение рассчитывают на базе вещества несодержащего летучих. П р и М е р 1. 22(Е)дОктен 150Белую суспензию фильтруют в горячем состоянии н растворитель отгоняют в вакууме. Результирующую смолу растирают с диэтиловым эфиром для удаления избыточного 3 иодопропанола. Остаточный растворитель отгоняют в вакууме с получением аморфного твердого вещества, которое по данным ЖХВДанализа определяют как 63/-трансЦис-четвертичную иодндную смолу, Материал растворяют в НО (45 мл), охлаждают до ОС и фильтруют для удаления осажденного цис-изомера. Преобч разование в хлоридную соль осуществляют пропусканием обогащенных трансизомеров жидкостей через колонку, набитую ионообменной смолой (35 г). Элюант концентрируют под вакуумом. Растирание остаточного масла с ацетотиджтшюшщшюсшъввщюбшюю твердого вещества. Суспендированиеи твердого вещества вдсухом Ыты-диметилформамшде (20 мл) при 8 ОС в течение Ю мин удаляют последние сшеды цисизомера. Материал суспеидируют в горячем ацетоне для удаления остаточного ЫЫ-диметипформамда и фильтруют с получением 4,8 г (842) четвертичного хлорида, которы по данным ЖХВДанализа определяется как 00 пый трансиэомер тпл 212-213 С. дПодтверждение транс-ориентации даетрентгеновскнй кристаллографический анализ перхлоратной соли соеди нения А.(500 мл) Растворитель отгоняют в ваг кууме Результирующую смолу растирают с диэтипоым эфиром для удаления из лишнего 3 нодопропанола. Остаточный растворитель ОТГОНЯЕРТ В ВЗКУУМЕ С ПО Пучением оранжевого масла, которое(200 мл) иохлаждают при -5 С в те говещества, 5 чение 8 ч Твердое вещество отфильтровывают и сушат втченне 2 ч при 50 С С получением 37,1 г четвертичноиодидной смеси (3 трансцнс по данным ЖХВД-анализа). Твердое вещество растирают с водой (200 мл) и отфильтровывают с получением 10,5 г очищенного цис-четвертичного нодида. Перекристаллизация из ацетонитршта дает 7,7 г (112) белого, кристаллического твердого вещества, топл. 1421 ц 4 сПодтверждение цисториентацин получено по даннм.рентгеновского крист таллографического анализа соедмне ния В.транс-Ы-3-Гидроксипропил-5-мет окснлауданозинийтхлорид (1002 транс по данным ЖХВД, 5,9 п)сУспендирушт в 80 мл 1,2-дихлорэтана при 7 ОС. добавляют 1,2 г хлорангидрида СЕ)-4-октен,8 тдикарбоновой кислоты (из вестное соедннение)-и смесь перемешивают 72 ч при температуре окружают щей среды. Реакционную смесь фильт-. РУЮТ Н РЗСТВОРНТЕПЬ ОТГОННЮТ В Ва кууме с получением аморфного твердокоторое суслендируютВ НОМ ВОДНОМ РЗСТВОРЕ ХЛОРИСТОГОЙ натрия суспензию доводят до рН 8,0 12-ной гидроокисью натрия и экстрагируют хлороформом (320 О мл). Скомбинированные хлороформовые порции ОСУШЗЮТ над БЕЗВОДНЫМ ХЛОРИСТЫМ КНЛЬдо сухого остатка. Остаток растворяют в 100 мл зтанолаи упаривают до образованя пены, которуюзатем упаривают до постоянной массы в условиях высокого вакутма (0,5 мм рт.стдД Выявлено, что белое тверд 0 г ВеЩест во (2,8 г, 41) имеет иистоту 952 по данным ЖВД. .,транс-Ыт 3 тГидроксипропцп-5-метоксилауданозннийхлорид (соединение А, 100 транс по данны ЖХВД, 5,88 Ы суспеиднруют в 80 мл 3,2-дихлорэтана при 70 С И добавляют хлорангидрид(Е)-4-октен-1,8 днкарбоновой кисло-ты (1,2 г). Хпорангидрид дикарбоновой.кислоты получают аналогично прит меру 1. Смесь перемешвают при температуре окружавшей среды в течение 16 ч И фильтруют. Фильтрат концентрируют до пены и делят между водой(65.мл)и ннтрометаном-(15 мл). Воднуш порцию промывают днэтновы эфн 5 ром и обрабатывают хлористым натрием(1,б г). Раствор в рассоле экстраги-руют хлороформом (40 мл), Хлороформовый экстракт концентрируют досмо лы А впоследствии растворяют в 2,5 ном водном растворе хлористого натрия (65 мл)- добавлением 0,1 М гидроокисннатрия рН доводят до 9,0 и водны раствор экстрагируют хлороформам (40 мл). Хлороформовый раст вор промвают-5 нм водным хлоридом натрия (200 ьш), осушают-вед безвод-ч ны хлоридом кальция, фильтруют и С упаривают.до сухого остатка при пониженном давлениик Твердое вещество растворяют в этаноле (952) и упаривают до пены припониженном давлении. Пану доводят до постоянной массы в условиях вакуума 0,5 мм рт.ст. с получением 4,0 г 57 соединенияз ил) изохинолнний дихлорид Середине ние С)(46,д г) в метаноле (240 мл) добавляют дибензоилвинной кислоты моногидрат (45,2 Г). Смесь еагреваютдо кипения, охлаждают при ЬС В теченне 16 ч и дибензоиптартрат (5)- 5-метоксипауданозиний (3 Е,6 г, 80) отфильтровывают и отбрасывают МаточНЫЭ РЭСТВОРЫ Подщепачивают концентРнрованной водой ЫаОН и упаривают в вакууме. Твердый остаток делят междуН 20 (200 мл) и диэтиловым эфиром(2150 мл). Эфирную фазу осуюают и уваривают с получением масла (24,9 г) К маслу В метаноле 128 мл добавляютдИбензоилвинной кислоты моногидрат (26 Ь г. Смесь нагревают до кипения и охлаждают при 5 С в течение 16 ч. Кристаллы собирают и перекрнсталлизовывают из метанола, пока не достигнуто удельное вращение Ы 3177 (14 ЕЕОН). Выход дибензонлтартрата (Н)-(3-5-метоксилау данозиния в виде белых кристаллов составляет 29,4 г (66)Порцию соли(150 г) в метаноле (200 мл) подщелачивают концентрированной водной Ыа 0 Н Смесь упаривают в вакууме и остаток делит между но (200 мл) идиэтиловым эфиром (2 ж 200 мл). Скомбинированные эфирные слои осушают и упаривают в вакууме с получением 7,2 г (922) (К)-(-)-5-метоксилауДЗНОЗИНЗ В виде масла.(125 мл) в течение 16 ч. Белую суспензию фильтруют в горячем состоянии и растворитель удаляют из фильтрата в вакууме. Остеточную смолу растирают с горячим этилацетатом для удаления избыточного 3-иодопропанола,растворяют в 200 мл метанола и пропускают нерез колонку, набитую ионот обменной смолой (60 г хлоридной формы). Элюант подвергают отгонке растворителя в вакууме с получением четвертичногхлоридиой соли (8,д г) в виде аморфного твердого вещества. По данным ЖХВД-анализа определяют как смесь 2,3 (1-транс)цис-диастерео изомеров.(2,3/1-транс-цис по данным ЖХВЦ, 2,5 г) растворяют в 60 мл 1,2-ди- О хлорэтана при температуре около 70 С. Добавляют хлорангидрнд (Е)-4-октен 1,8-дикарбоновой кислоты (О,5 г) исмесь Перемешивают 19 ч при темпера туре окружающей среды. Растворитель Удаляют в вакууме с получением аморфного твердого вещества, которое растворяют в хлороформе (25 мл) и промывают 52-ным водным раствором хлорида натрия (3135 мл) для удаления непрореагировавшин четвертичных солей. Хлсроформовый слой осушают и упорнвают в вакууме с получением аморфного твердого-вещества. Примеси-слопг не эфиры кислоты удаляют промыванием горячим 2-бутаноном Остаточный растворитель упаривают в вакууме и ре. ЗУП-ЬТИРУПЦЕ аморфное. твердое веще ство растворяют В метаноле, фильтруют и лиофнлизуют с получением 1,9 г(Е)-(1 Н,1 К)-224-октендиоипбис(окситриметилен 1 бис,2 д 3-тетрагидро-67-диметокси-2-метил-1(345-триетоксибензил)изохинолиний дихлорида соединение С, которое по данным ЖХВД-анализа считается смесью 44,62 КБ-ВБ (транс-траНс)днэфирад д 24 ЕЛ-КБ (Цис-транс)днэфира, 7,5 взв (цис-цис) диэфира 4,0 ЕБ(транс) сложного эфира кислоты и 1,5 ЕД (цис) сложного эфира кисло ты. д-627 11,92 в ню).п р н м е р 4, Хроматографическое разделение отдельных компонентов соециненияДля проведения этого разделения испольеуют ЖХВД-систему Ыасегв НРЬС/зузсеш 5 О 0 А снабженную двумя сипикагельными кассетами сдвоеипого типе. Колонки предварительно УРНВНО вешивают в подвижной фазе(этанол метанолтетраметиламонийхлорид 6004001) и в колонку аагружают смесь Диэфиров соединение С (5 г)в этаноле (25 мл). Систему ГЭПЮИРУЮТ с ИСпОЛЬЗОВаНШЕМ 132 л подвижной фазы, которую собирают в виде 66 фракций (по 200 мл). Фракции еиализис руют с помощью аналитической ЖХВДи на основании этих данных определяют в комбинированном виде лак следующие соединенияФракции 26-30 комбинируют и унарнвают при пониженном давлении. Результирующнй остаток растирают с хлороформом (2 ОО мл) и фильтруют. Фильтрат промывают 52-ным водным хлоридом тнатрия и концентрируют при пониженном давлении с получением масла. Масло растворяют В этаноле-(50 мл) н уваривают до пены (0,4 г)БЩной фосфорной кислоте(0 мл) на гревают 18 ч при 60-7 ОС и анализируют с помощью ЖХВД. Наблюдается цисчетвертичная соль до исключенияЖДЭЕТСЯ СОВМЕСТНЪЫИ ИНЪЕКЦИНМИ С СОЕдиненнями А и В. Соединение 1. (Е)-(К,1 В, 2 Е 25)-2,2-Д-0 ктендиоил-бис(окснтри метиленП бис 1 2,3 о 4-тетрагидро-6 7 диметокси-2-метил-1-3,4,5-триметоксиб енз ил) изохинолинит днхпорнд . Фракции 34-36 комбинируют и продукт выделяют-аналогично соединению Н. Из них получают 2,0 г белой пены. Ен 54 о (152 н,о). 3 н ямг (свс 1,) или тмс 8 6,64Вычислено, С 61,28 Н 7,76 Н 2,36 с 1 6,01 с 5 анъ(111)ос 121 5 61,04 3 сн 50 н Найдено, 2 С 61,32 Н 7,71 Ы 2,36 с 1 5,97. Соединение 1 (10 мг) в 1-ной водной фосфорной кислоте (10 мл) на гревают при 60-70 С в течение 18 чИ анализируют с помощью ЖХВД. НаблЕг дают равные количества цис и трансчетвертичных солей. Это подтверждается совместными инъекциями с соединениями А и В. Соединение 3. (Е)(1 в 1 в, 25,25)224-Октендиоил-бис(окситриметиленп бис ж , дъ-д-тетрагидро-б ь 1668614Комбнннруют фракции 56-66 и продукт выделяют по методике выделения сощшнщшяЪ 1 ш тж шщштют 0,9 г не совсем белой пены -76,7Соединение 0 мг в 12-пой водной фосфорной кислоте (10 мл нагревают при 60-7006 в течение 1 ч и анализируют с помощью ЖХБД Наблюдается транс-четвертичная соль до исключения цис-четвертичной соли, Это подтверждается совместными инъекциям с соединениями А и ВыСоединения предлагаемойформулы являются мощныи нервно-мшшечноблокирующнми средствам, проявляющими относительно короткую продолжительностьдействИя составляющую например около КО мин при испытании на п обезьянах. Эти соединения характеризуются недеполяризующим механиамом действия, являются фармакологически обратимыми, обеспечивают относительно быстрое наступление действия, что является положительным эффектом, имеющим исключительно большое значениеСоединения предлагаемой формулы используются в качестве нервномышечно блокирующих средств при прове- ДЕНИИ ХЕИРУРГИЧЕСЕИХ операций ИДЕИ ИНтубации трахеи,для чего их вводят традиционны парентеральным путем,-например внутримншечным или внутри ванным вливанием.Предлагают также способ расслабления мускулатуры утелекопитающих, заключающейся в том, чтомекопнтающим вводят эффективно действующее нервно-мышечно блокирующее количество соединение предлагаемой формулы. Соединение предлагаемой . формулы может применяться в медицине или ветеринарии, особенно для расслабления мускулатуры у млекопитающих. Соединения вводят субъектам,таким как обезьявътнли людям, идру
МПК / Метки
МПК: C07D 217/10, A61K 31/47
Метки: изохинолиния, получения, производных, способ
Код ссылки
<a href="https://by.patents.su/8-1329-sposob-polucheniya-proizvodnyh-izohinoliniya.html" rel="bookmark" title="База патентов Беларуси">Способ получения производных изохинолиния</a>
Способ получения производных фенилгуанидина и производные сульфокислоты в качестве промежуточных продуктов в синтезе производных фенилгуанидина

Номер патента: 937
Опубликовано: 15.12.1995
Авторы: Ференц Шпербёр, Ева Шомфаи, Чаба Хусар, Аттила Немет, Лайошне Пали, Петер Шаркёзи
МПК: A61K 31/185, C07C 317/42, C07C 309/15...
Метки: получения, качестве, продуктов, способ, фенилгуанидина, промежуточных, производных, сульфокислоты, производные, синтезе
Текст:
...смеси.Амин общей формулы (Ш), преимущественно, используют в избытке 580.Согласно изобретению, преимущество способа в том, что он прост и не приводит к получению пахнущих или токсичных побочных продуктов, а выделенные полупродукты стабильны при комнатной температуре. Процесс идет с хорошим выходом (около 70) при незначительном времени реакции.Соединения общей формулы (П) и НУ) известны в практике и могут быть получены хорошо известными...
Способ получения производных имидазола

Номер патента: 336
Опубликовано: 30.12.1994
Авторы: Джон Джонас Витаутас Дансиа, Панкрас Чор Бун Вонг, Дэвид Джон Карини
МПК: A61K 31/415, C07D 233/58
Метки: имидазола, получения, способ, производных
Текст:
...колоночная хроматография (злюация 0-51, метанола/хлороформ) приводит к 0,33 г 1 СТ-карбометоксибифенил-дищметилН-бутил-Е-оксиметилимидазола.Следующие промежуточные продукты,приведенные в табл. 1 также получены по методикам, описанным в примере 1.Раствор 5 г 1 Юдкарбметоксидифенид 4-ил)метил-2-6 утил 4-хлор-Б-оксиметилимидазола И 1 мл концентрированной серной кислоты в 200 мл метанола кипятят с обратным холодильником в течение 20 ч. После...
Способ получения производных 3,5-диамино-1,2,4-триазина или их кислотно-аддитивных солей
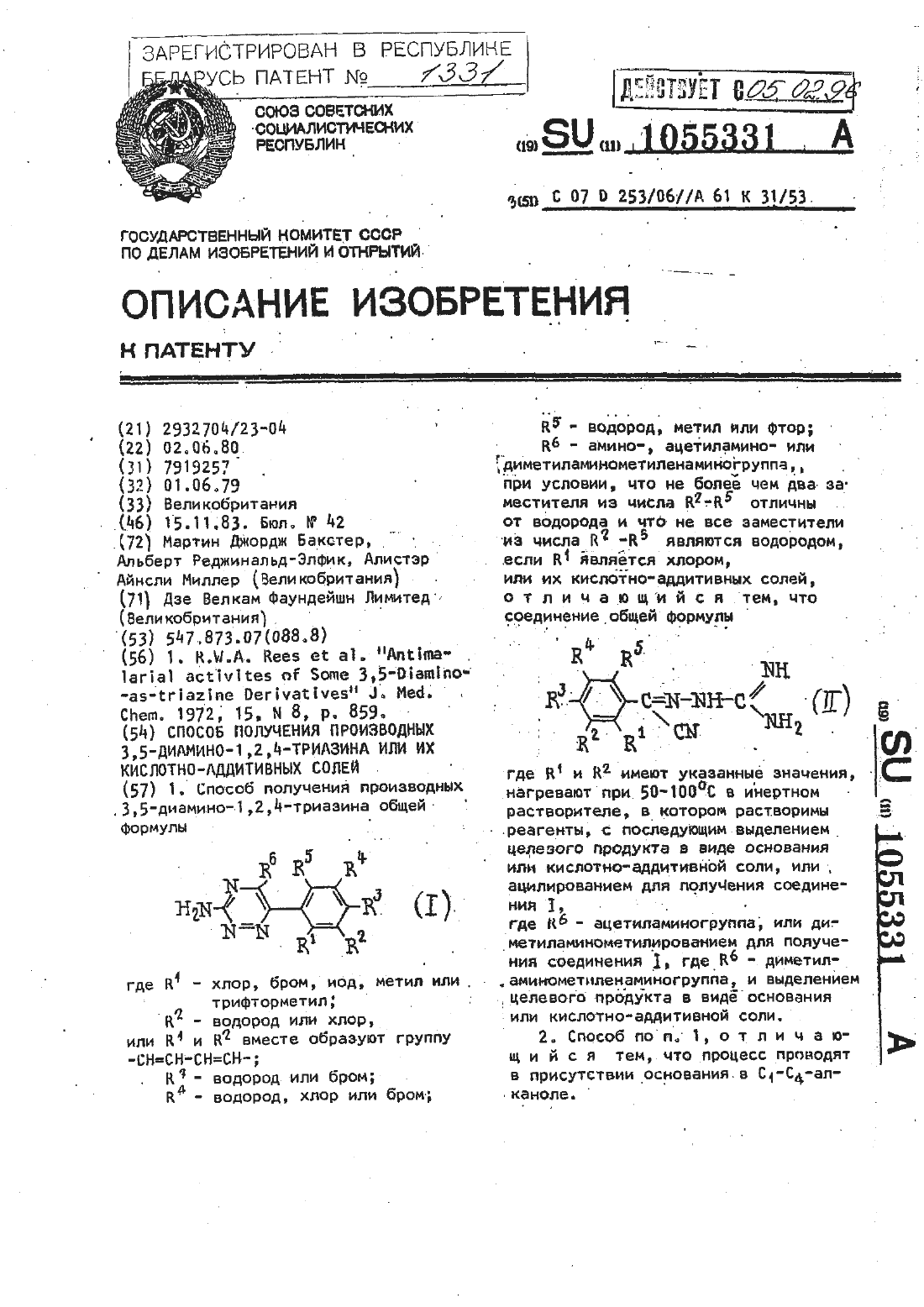
Номер патента: 1331
Опубликовано: 16.09.1996
Авторы: Альберт Реджинальд-Элфик, Мартин Джордж Бакстер, Алистэр Айнсли Миллер
МПК: A61K 31/53, C07D 253/06
Метки: получения, солей, способ, производных, 3,5-диамино-1,2,4-триазина, кислотно-аддитивных, или
Текст:
...прикомнатной температуре в течение 2 ч, затем его медленно добавляютв перемешиваемый раствор хлорида одновалентной меди (97,0 г, 0,97 М)в концентрированной соляной кислоте 5(970 мл) Полученную смесь перемешивают до тех пор, пока не прекратится выделение азота, а затем выдерживают в течение ночи. Твердое вещество отделляют фильтрацией, промывают водой и. 10 сушат в вакууме, Выход 90,1 г (892),т.пль 160-1 б 5 ЧС.(200 мл )нагревают до...
Способ получения сульфинильных производных гетероциклических соединений или их фармацевтически приемлемых солей

Номер патента: 77
Опубликовано: 30.09.1994
Авторы: Дэвид Кокс, Джон Льюис Сучитский, Энтони Говард Инголл
МПК: C07D 263/58, A61K 31/415, C07D 235/28...
Метки: приемлемых, фармацевтически, гетероциклических, сульфинильных, или, соединений, способ, производных, получения, солей
Текст:
...25 меру 1 а. 7 - 3(1 Н 2 Бензимидазолипсульфиннл метил)-ЫПдиметил 2 пиридинамин (е)г Продукт ц превращают в целевое соединение (т.пл. 12 д 126 С)по прн- за меру 1 Ь П р им е р 5. 2-(Н 2 Бензиидазолилсульфинилметил)дбенволамнн. Ы 2 дХлорметнлфенил 32 д,6 триметилбенволсульфонамнд (а). Ы(2 Гидроксиметилфенил)-24,6-триметилбенэопсулЬфонамид(40 Г) в сухом дихлорэтана (80 мл) обрабатывают тионипхлоридом 01,15 ил) ПРИ КОМ натной температуре...
Способ получения метиленовых производных андроста-1,4-диен-3,17-диона
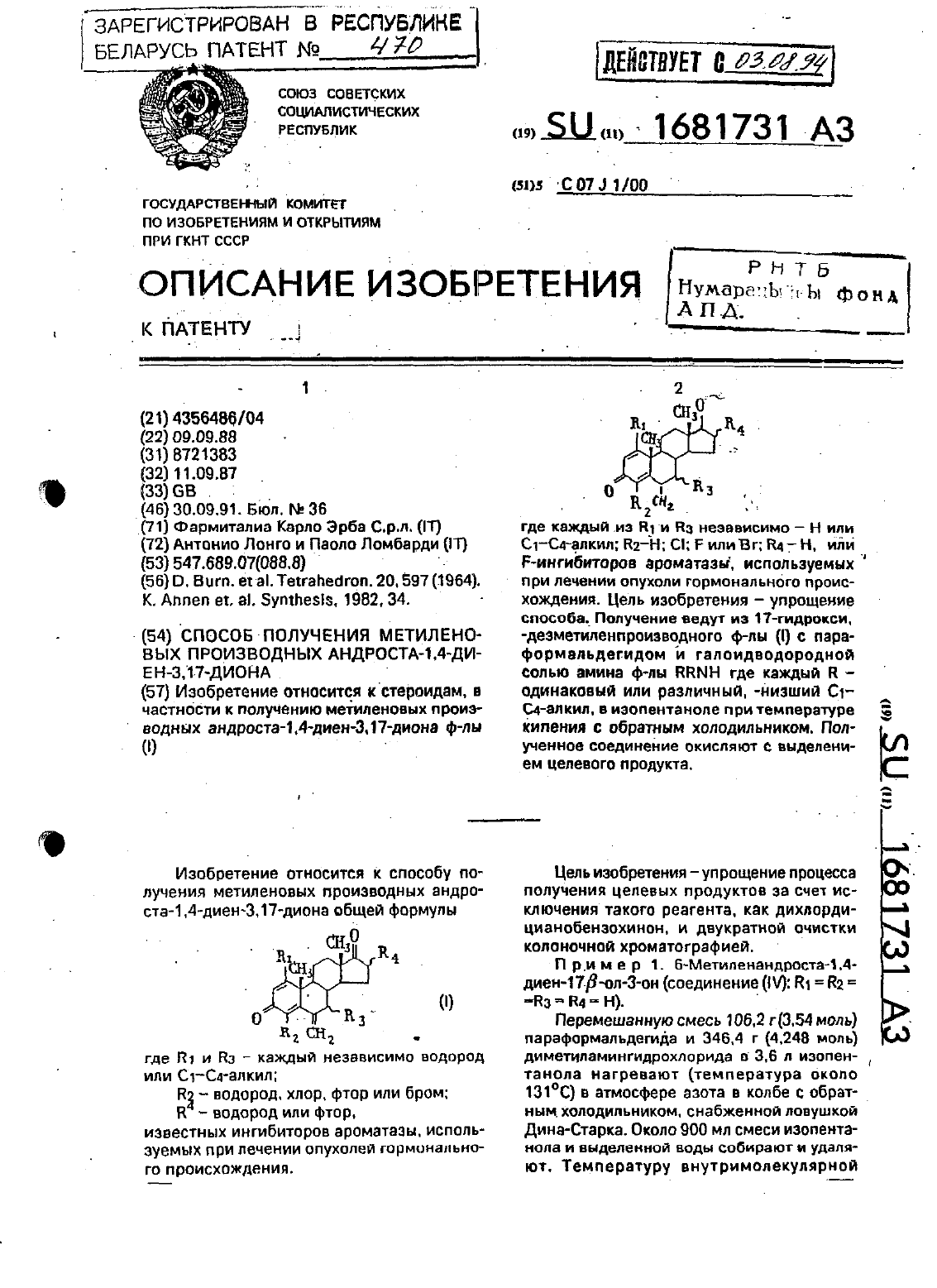
Номер патента: 470
Опубликовано: 30.03.1995
Авторы: Паоло Ломбарди, Антонио ЛОНГО
МПК: C07J 1/00
Метки: получения, андроста-1,4-диен-3,17-диона, метиленовых, способ, производных
Текст:
...смеси этанола и воды (7030). Отфипьтрованный белый осадок счшат в ваку уме при 40 С. получая 283 г (0963 моль,выход 30.7 Ы данного продукта. Т. пл. 135137 С. СК перемешанному раст-вору 2.8 г (0963 моль) Б-метиленандростач д-диен-1 ЦЗ З-она в 700 мл ацетона при 4109 Сд 0 бавляют по каплям 35 мл реагента джонса. После этого реакционную смесь перемешивают в течение 10 мин и затем тщательно обрабатывают 50 мл изопропанола После дополНИТВЛЬНОГО...
Предыдущий патент: Гербицидная композиция (ее варианты)
Следующий патент: Способ получения 3-[2-диметиламино)-этил]-N-метил-1Н-индол-5-метансульфонамида или его солей, или сольватов
Случайный патент: Внутрисхемный эмулятор