Производные эритромицина, способ их получения, фармацевтическая композиция на их основе и промежуточные соединения
Номер патента: 4746
Опубликовано: 30.12.2002
Авторы: ДЕНИ, Алексис, ГУЭН Д'АМБРИЕР, Солянж, АГУРИДАС, Константин, ЛЕ МАРТРЕ, Одиль, ШАНТО, Жан-Франсуа


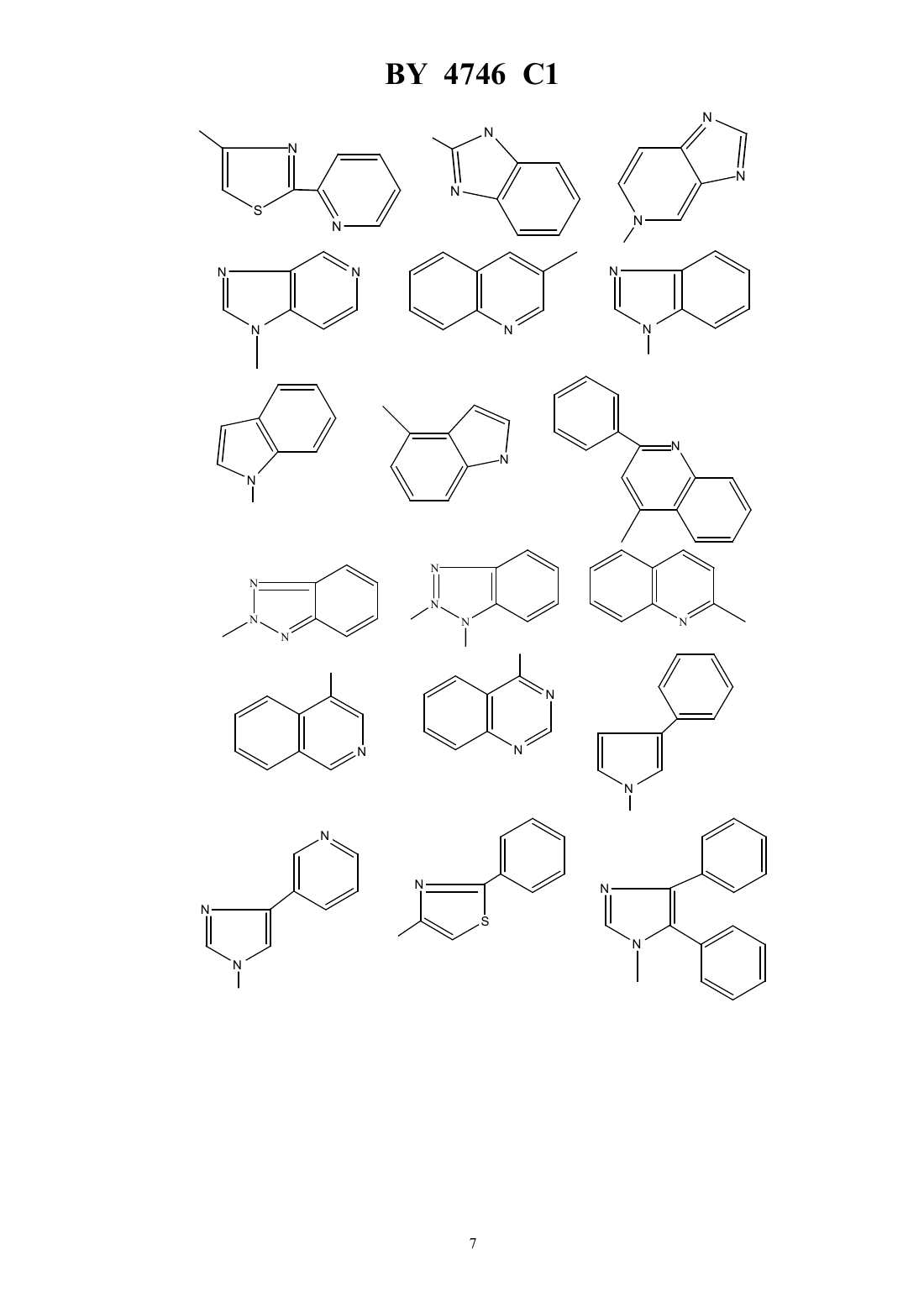

Текст
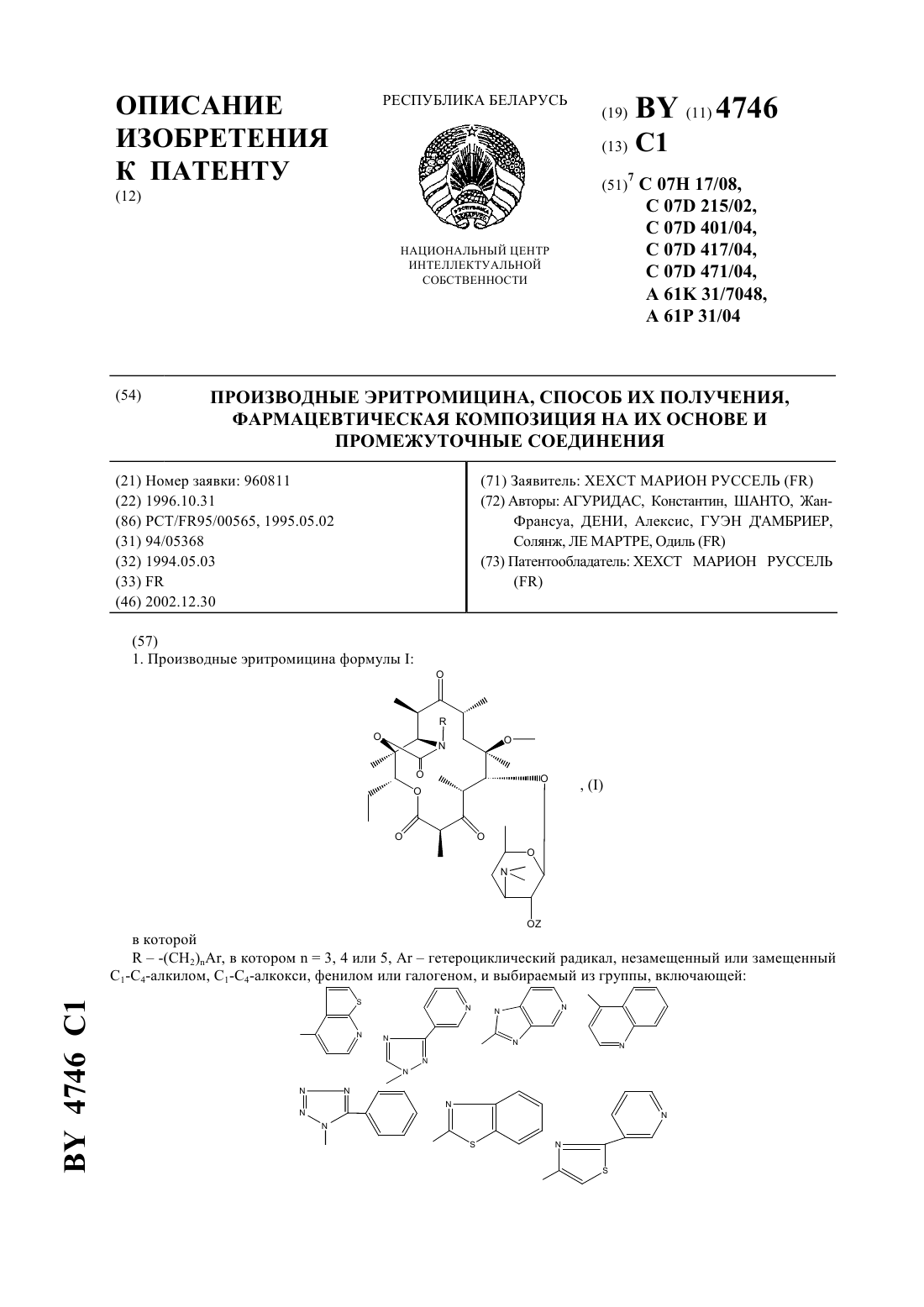
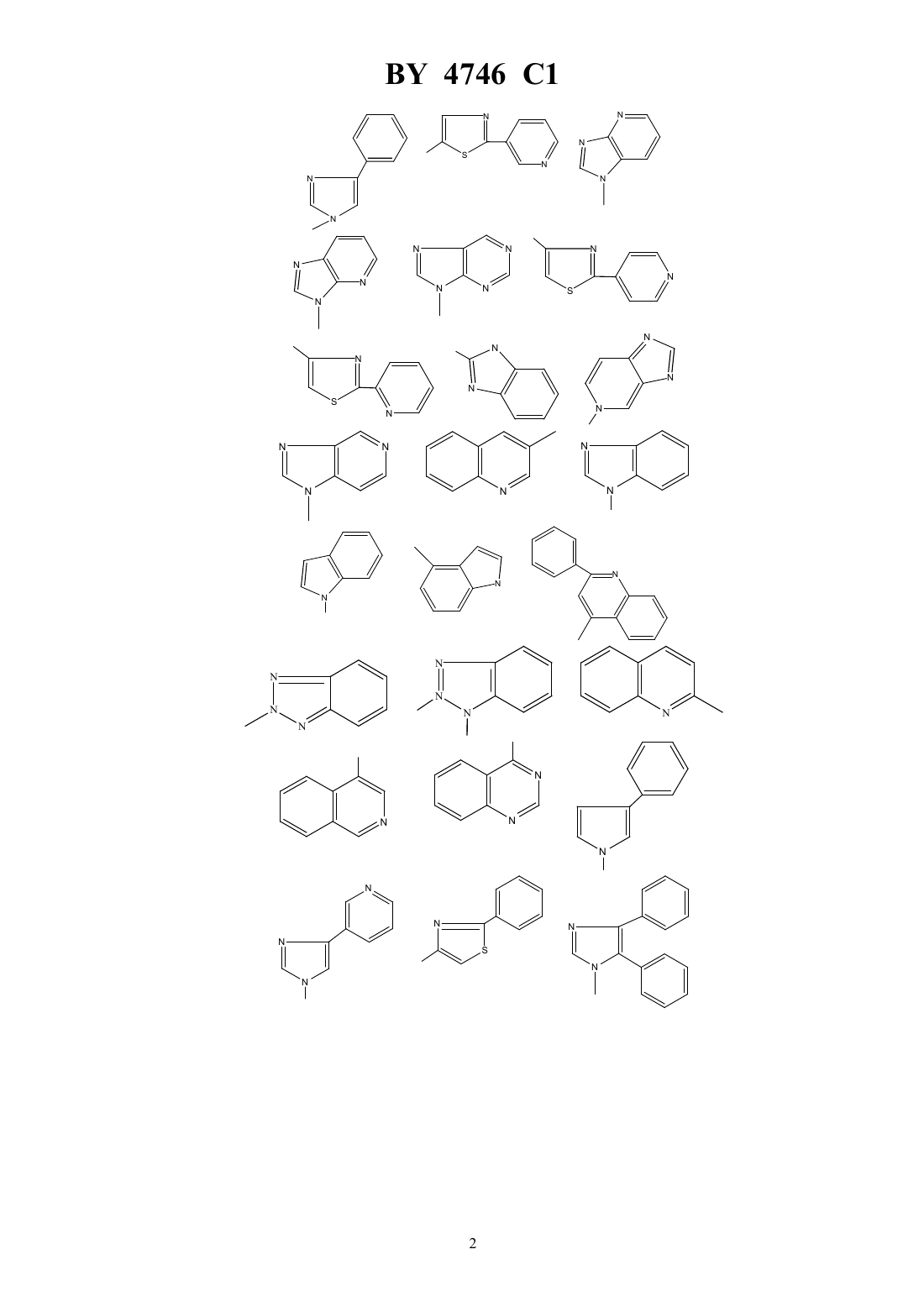
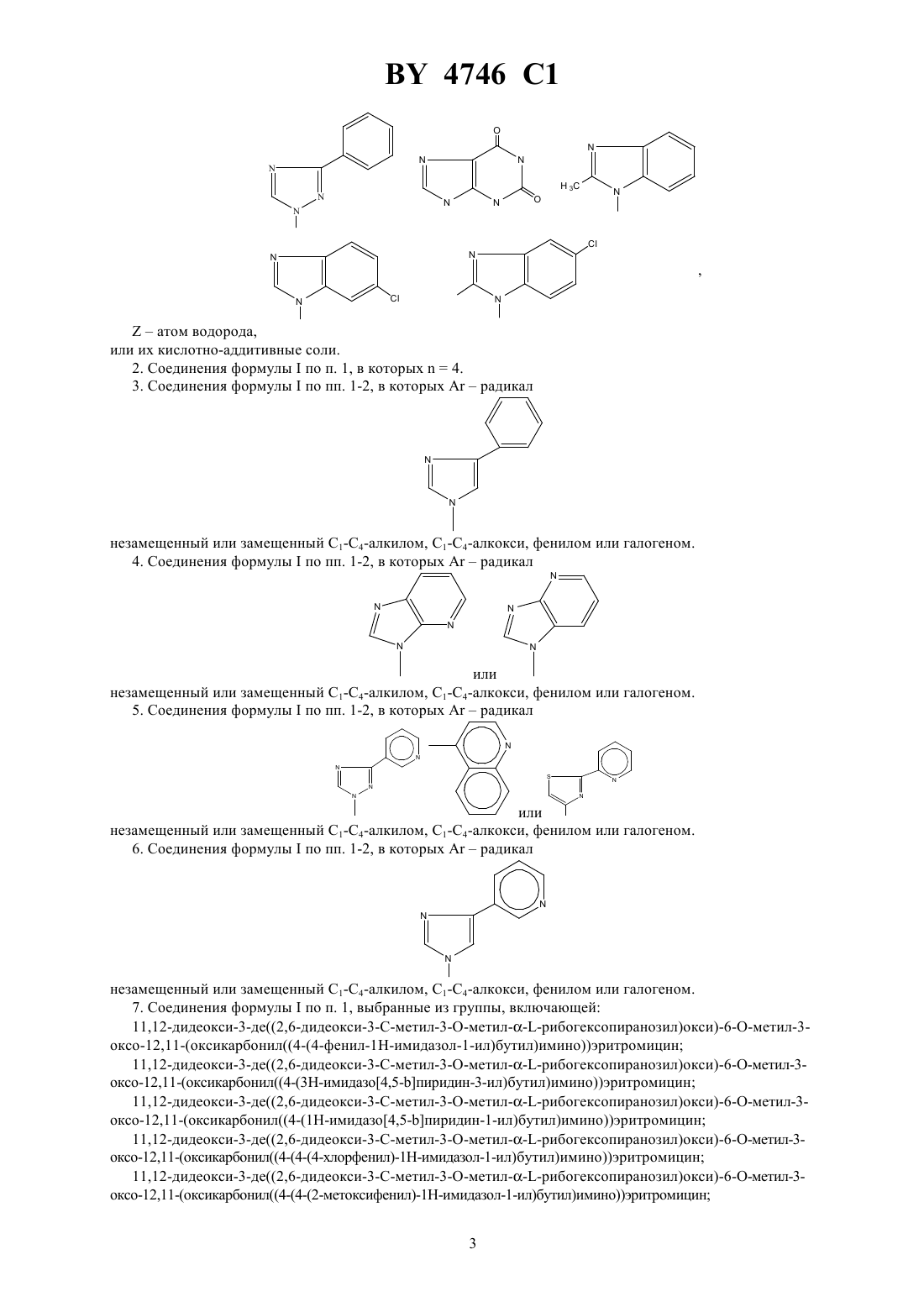
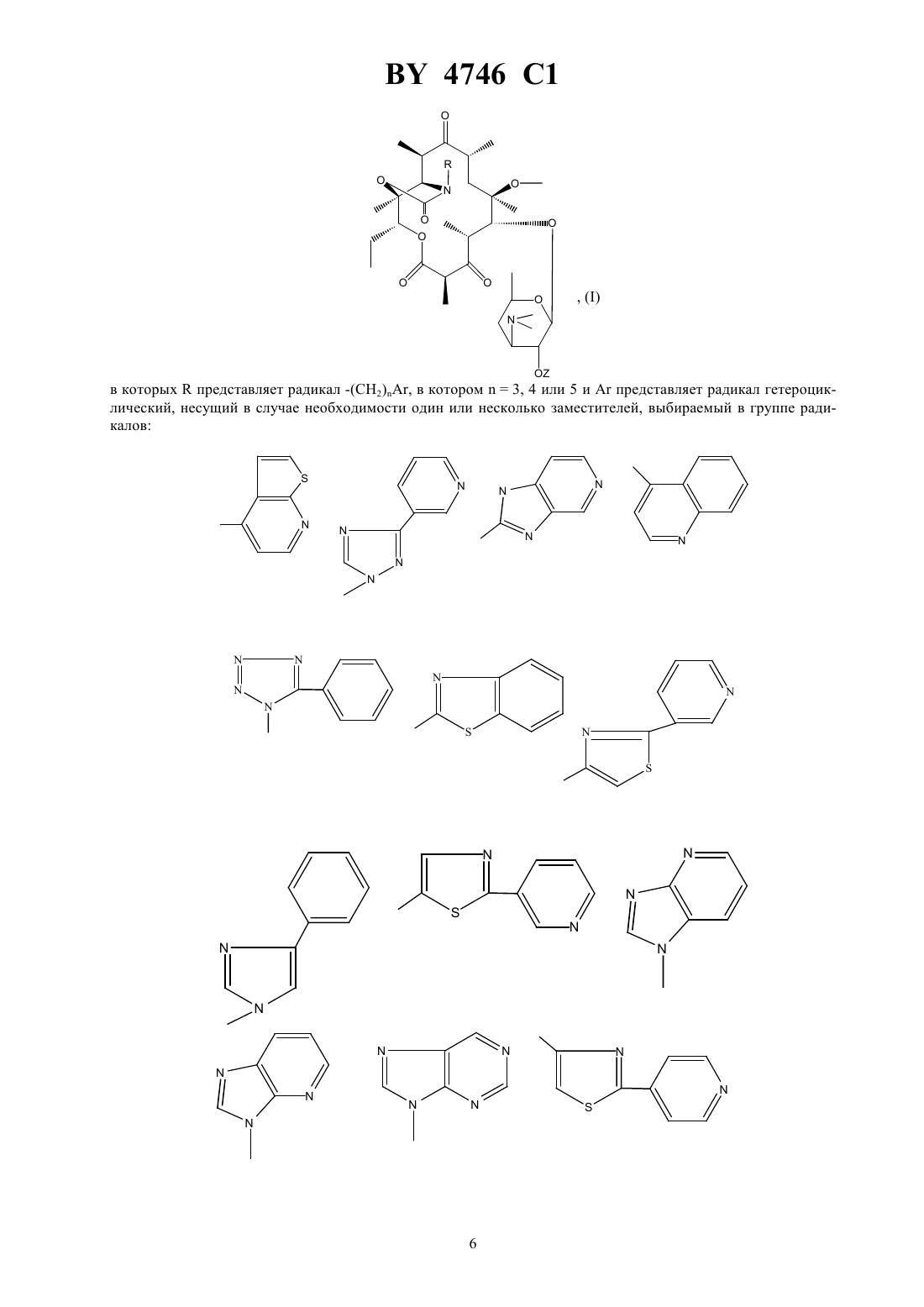
НАЦИОНАЛЬНЫЙ ЦЕНТР ИНТЕЛЛЕКТУАЛЬНОЙ СОБСТВЕННОСТИ ПРОИЗВОДНЫЕ ЭРИТРОМИЦИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ,ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ(71) Заявитель ХЕХСТ МАРИОН РУССЕЛЬ(73) Патентообладатель ХЕХСТ МАРИОН РУССЕЛЬ(57) 1. Производные эритромицина формулы в которой-(2), в котором 3, 4 или 5,гетероциклический радикал, незамещенный или замещенный 1-4-алкилом, 1-4-алкокси, фенилом или галогеном, и выбираемый из группы, включающей незамещенный или замещенный 1-4-алкилом, 1-4-алкокси, фенилом или галогеном. 4. Соединения формулыпо пп. 1-2, в которыхрадикал или незамещенный или замещенный 1-4-алкилом, 1-4-алкокси, фенилом или галогеном. 5. Соединения формулыпо пп. 1-2, в которыхрадикал или незамещенный или замещенный 1-4-алкилом, 1-4-алкокси, фенилом или галогеном. 6. Соединения формулыпо пп. 1-2, в которыхрадикал незамещенный или замещенный 1-4-алкилом, 1-4-алкокси, фенилом или галогеном. 7. Соединения формулыпо п. 1, выбранные из группы, включающей 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метилрибогексопиранозил)окси)-6 метил-3 оксо-12,11-(оксикарбонил 4-(4-фенил-1-имидазол-1-ил)бутил)иминоэритромицин 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метилрибогексопиранозил)окси)-6 метил-3 оксо-12,11-(оксикарбонил 4-(3-имидазо 4,5-пиридин-3-ил)бутил)иминоэритромицин 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метилрибогексопиранозил)окси)-6 метил-3 оксо-12,11-(оксикарбонил 4-(1-имидазо 4,5-пиридин-1-ил)бутил)иминоэритромицин 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метилрибогексопиранозил)окси)-6 метил-3 оксо-12,11-(оксикарбонил 4-(4-(4-хлорфенил)-1-имидазол-1-ил)бутил)иминоэритромицин 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метилрибогексопиранозил)окси)-6 метил-3 оксо-12,11-(оксикарбонил 4-(4-(2-метоксифенил)-1-имидазол-1-ил)бутил)иминоэритромицин 3 4746 1 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метилрибогексопиранозил)окси)-6 метил-3 оксо-12,11-(оксикарбонил 4-(4-(4-фторфенил)-1-имидазол-1-ил)бутил)иминоэритромицин 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метилрибогексопиранозил)окси)-6 метил-3 оксо-12,11-(оксикарбонил 4-(7-метокси-4-хинолинил)бутил)иминоэритромицин 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метилрибогексопиранозил)окси)-6 метил-3 оксо-12,11-(оксикарбонил 4-(2-(2-пиридинил)-4-тиазолил)бутил)иминоэритромицин и 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метилрибогексопиранозил)окси)-6 метил-3 оксо-12,11-(оксикарбонил 4-(3-(3-пиридинил)-1-1,2,4-триазол-1-ил)бутил)иминоэритромицин. 8. Соединение формулыпо п. 1, представляющее собой 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метилрибогексопиранозил)окси)-6 метил-3 оксо-12,11-(оксикарбонил 4-(4-(3-пиридинил)-1-имидазол-1-ил)бутил)иминоэритромицин. 9. Соединения формулыпо любому из пп. 1 - 6 или их фармацевтически приемлемые кислотноаддитивные соли, обладающие антибактериальной активностью. 10. Соединения формулыпо п. 7 или их фармацевтически приемлемые кислотно-аддитивные соли, обладающие антибактериальной активностью. 11. Соединение формулыпо п. 8 или его фармацевтически приемлемые кислотно-аддитивные соли, обладающие антибактериальной активностью. 12. Фармацевтическая композиция, обладающая антибактериальной активностью, содержащая в качестве активного ингредиента терапевтически эффективное количество соединения по любому из пп. 9-11 и фармацевтически приемлемый носитель. 13. Способ получения соединений формулыпо п. 1, заключающийся в том, что соединение формулы в которой- остаток карбоновой кислоты, содержащий до 18 атомов углерода,подвергают взаимодействию с соединением формулы 2 ,в которойимеет значения, указанные в п. 1,с получением соединения формулы в которойиимеют указанные выше значения,которое в случае необходимости подвергают гидролизу по положению 2 и/или в случае необходимости действию кислоты с получением соли.(56)2692579 1, 1993.0487411 1, 1992.0596802 1, 1994. Изобретение относится к новым производным эритромицина, к способу их получения, к фармацевтической композиции на их основе и к промежуточным соединениям. Изобретение относится, в частности, к соединениям формулы в которыхпредставляет радикал -(2), в котором 3, 4 или 5 ипредставляет радикал гетероциклический, несущий в случае необходимости один или несколько заместителей, выбираемый в группе радикалов,ипредставляет атом водорода или остаток кислоты, а также к их солям присоединения кислот. В качестве примера солей присоединения производных формулыс минеральными или органическими кислотами можно назвать соли, образованные с кислотами уксусной, пропионовой, трифторуксусной, малеиновой, винной, метансульфокислотой, бензолсульфокислотой, паратолуолсульфокислотой, соляной, бромистоводородной, йодистоводородной, серной, фосфорной и особенно с кислотами стеариновой, этилянтарной или лаурилсерной. Гетероциклический радикал может быть замещен одним или несколькими радикалами, выбираемыми из группы,образованной 1-6-алкилом, 1-6-алкоксилом, трифторметокси, галоидом. Когда гетероциклический радикал содержит несколько циклов (связанных между собой или конденсированных), один или несколько заместителей могут находиться на одном и/или на другом из циклов, гетероциклических или карбоциклических так, например, если гетероциклическое кольцо соединено или конденсировано с радикалом арила, то гетероциклическое кольцо и кольцо арила могут оба нести один или несколько заместителей. Радикал алкил представляет предпочтительно радикал метил, этил, пропил, изопропил, н-бутил, изобутил, трет-бутил,галоген представляет предпочтительно фтор, хлор или бром,остаток карбоновой кислоты представляет предпочтительно остаток ацетила, пропионила, бутирила, изобутирила, н-валерила, изовалерила, трет-валерила и пивалила. Изобретение касается особенно соединений формулы , в которойпредставляет собой атом водорода,и соединений формулы , в которойравно числу 4. Более конкретно изобретение касается соединений формулы , в которойпредставляет радикал в случае необходимости замещенный, а также соединений формулы , в которыхпредставляет радикал в случае необходимости замещенный, а также соединений формулы , в которыхпредставляет радикал или ,или в случае необходимости замещенный, и особенно соединений формулы , в которыхпредставляет радикал в случае необходимости замещенный. Изобретение относится особенно к соединениям формулы , получение которых описано ниже в экспериментальной части. Среди полученных соединений изобретения можно назвать следующие соединения 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(4-фенил-1-имидазол-1-ил)бутил)иминоэритромицин 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(3-имидазо(4,5-)пиридин-3-ил)бутил)иминоэритромицин 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(1-имидазо(4,5-)пиридин-1-ил)бутил)иминоэритромицин 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(4-(4-хлорфенил)1-имидазол-1-ил)бутил)иминоэритромицин 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(4-(2-метоксифенил)1-имидазол-1-ил)бутил)иминоэритромицин 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(4-(4-фторфенил)1-имидазол-1-ил)бутил)иминоэритромицин 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(7-метокси-4-хинолинил)бутил)иминоэритромицин 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(2-(2-пиридинил)4-тиазолил)бутил)иминоэритромицин 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(3-(3-пиридинил)1-1,2,4-триазол-1-ил)бутил)иминоэритромицин и особенно 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(4-(3-пиридинил)1-имидазол-1-ил)бутил)иминоэритромицин. Продукты общей формулыимеют очень высокую антибиотическую активность, в отношении таких грамположительных бактерий, как стафилококки, стрептококки, пневмококки. Соединения, согласно изобретению, можно, следовательно, использовать в качестве медикаментов при лечении инфекций чувствительных к медикаментам, в частности стафилококковых инфекций, таких как стафилококковый сепсис, злокачественные стафилококки лица или кожи, пиодермита, септические или гнойные раны, фурункулы, карбункулы, флегмоны, рожистые воспаления и угри, таких стафилококковых заболеваний, как первичные острые или с послегриппозные ангины, бронхопневмония, гнойные легочные, и стрептококковых инфекций, таких как острые ангины, отиты, синуситы, скарлатина, пневмококковых, таких как пневмония, бронхит бруцеллез, дифтерия, и гонококковых инфекций. Соединения настоящего изобретения являются также активными против инфекций, вызванных микробами, ,, , , ,или микробами типа , ,и . Соединения формулы , а также их соли присоединения фармацевтически приемлемых кислот могут использоваться в качестве антибиотиков. 4746 1 В качестве таких антибиотиков можно, например, использовать предпочтительные соединения формулы, указанные выше, в частности соединения примеров 1, 2, 3 и 29-35, а также их фармацевтически приемлемые соли. Изобретение касается также фармацевтических композиций, содержащих в качестве действующего начала, по меньшей мере, одно из определенных выше соединений. Эти композиции можно вводить оральным, ректальным, парентеральным путем или наносить локально на кожу и на слизистые оболочки, но предпочтительным методом введения является оральный путь. Они могут быть твердыми или жидкими и иметь фармацевтические формы, используемые обычно в медицине, например простые таблетки или драже, желатиновые капсулы, гранулы, свечи, препараты для инъекций, мази, кремы, гели их готовят обычными методами. Действующее начало можно вводить в композиции вместе с эксципиентами, применяемыми обычно в этих фармацевтических составах, такими как тальк,гуммиарабик, лактоза, крахмал, стеарат магния, масло какао, водные или неводные носители, жирные вещества животного или растительного происхождения, парафиновые производные, гликоли, различные смачивающие агенты, диспергаторы или эмульгаторы и консерванты. Эти композиции могут также выпускаться в форме порошка, годного для растворения по мере необходимости в соответствующем растворителе, например в апирогенной стерильной воде. Вводимая доза изменяется в зависимости от вида заболевания, от пациента, от метода введения и от используемого соединения. Она может составлять, например, от 50 мг до 300 мг в день при оральном введении взрослому пациенту соединения примера 1 или примера 2. Изобретение касается также способа получения соединений формулы , заключающегося в том, что соединение формулы в которомпредставляет остаток кислоты, подвергают взаимодействию с соединением формулы 2,в которойопределен выше, с получением соединений формулы в которыхиимеют указанное выше значение, затем полученное соединение формулы , в случае необходимости, подвергают действию агента высвобождения функции гидроксила в положении 2 и/или, в случае необходимости, действию кислоты для образования соли причем реакцию соединения формулыс соединением формулыпроводят в таком растворителе,как, например, ацетонитрил, диметилформамид или тетрагидрофуран, диметоксиэтан или диметилсульфоксид гидролиз функции сложного эфира в положении 2 осуществляют при помощи метанола или водной соляной кислоты и солеобразование осуществляют с помощью кислот по классическим методам. Соединения формулы , используемые как исходные продукты, описаны и заявлены в европейской заявке на патент 0596802. 10 4746 1 Соединения формулы 2 представляют собой в общем известные продукты, однако конкретные соединения, используемые для получения соединений в примерах заявки, являются новыми и сами составляют объект изобретения, и их получение описано ниже. Соединения формулы 2 можно получать, например, способами, описанными в . . . (1982), том 25, стр. 947 и следующие, том 32,14, стр. 1699-1702 (1991) . . . 54 (18) 4298, 301 (1989) . . . 28 (101) 2589 91 (1963) или патент ФРГ 3406416 6-895-901 (1941) или . . 17 (14) 1741-8 (1987). Более конкретно изобретение касается аминов формулы , определенной выше, получение которых подробно изложено ниже. В частности, изобретение относится к следующим аминам 4-фенил-1-имидазол-1-бутанамин,3-имидазо(4,5-)-пиридин-3-бутанамин,1-имидазо(4,5-)-пиридин-1-бутанамин,2-фенил-4-хинолинбутанамин,1-бензотриазол-1-бутанамин,2-бензотриазол-2-бутанамин,1-метил-1-имидазо(4,5-)-пиридин-2-бутанамин,3-метил-3-имидазо(4,5-)-пиридин-2-бутанамин,5-хлор-1-бензимидазол-1-бутанамин,7-метокси-4-хинолинбутанамин,1-имидазо(4,5-)-пиридин-1-бутанамин,9-пурин-9-бутанамин,1-метил-1-индол-4-бутанамин,3-фенил-1-1,2,4-триазол-1-бутанамин(хлоргидрат),5-фенил-1-тетразол-1-бутанамин(хлоргидрат),2-бензотиазолбутанамин,4-(тиено(2,3-)-пиридин-4-ил)бутанамин,5,6-диметил-1-бензимидазол-1-бутанамин,3-хинолинбутанамин,2-хинолинбутанамин,5-имидазо 4,5-пиридин-5-бутанамин,1-метил-1-бензимидазол-2-бутанамин,6-хлор-1-бензимидазол-2-бутанамин,2-метил-1-бензимидазол-2-бутанамин,4-(4-хлорфенил)-1-имидазол-1-бутанамин,2-(3-пиридинил)тиазол-5-бутанамин,7-метоксихинолин-4-бутанамин,4-(4-фторфенил)-1-имилазол-1-бутанамин,4-(2-метоксифенил)-1-имидазол-1-бутанамин,3-(3-пиридинил)-1-1,2,4-триазол-1-бутанамин,4-(3-пиридинил)-1-имидазол-1-бутанамин,2-(2-пиридинил)тиазол-4-бутанамин,2-фенилтиазол-4-бутанамин,4-(4-метоксифенил)-1-имидазол-1-бутанамин,изохинолин-4-бутанамин,хиназолин-4-бутанамин,4,5-дифенил-1-имидазол-1-бутанамин,4-(3-метоксифенил)-1-имидазол-1-бутанамин,4-(4-(трифторметокси)фенил)-1-имидазол-1-бутанамин,1,2,3,6-тетрагидро-1,3-диметил-2,6-диоксо-7-пурин-7-бутанамин,2-(4-пиридинил)тиазол-4-бутанамин,1-индол-1-бутанамин,2-(3-пиридинил)тиазол-4-бутанамин,а также их соли присоединения с кислотами. Пример 1. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(4-фенил-1-имидазол-1-ил)бутил)иминоэритромицин. 4746 1 Нагревают до 63 смесь 0,705 г продукта 2-ацетат-11-деокси 10,11-дидегидро-3-де 2,6-дидеокси-3-метил-3 метил-альфарибогексопиранозил)окси)-121-имидазол-1-ил)карбонил)-6 метил-3 оксоэритромицина (полученного, как указано в примере 1 европейской заявки на патент 0596802) в 3 мл ацетонитрила, содержащего 10 воды, и 1,08 г 4-(4-фенил-1-имидазол-1-ил)бутанамина. Поддерживают реакционную смесь при этой температуре в течение 5 часов. Оставляют до возвращения к комнатной температуре, выливают реакционную смесь в раствор кислого фосфата натрия, экстрагируют этилацетатом. Промывают органические фазы водой, сушат их, фильтруют и концентрируют. Получают 1,5 г продукта, к которому добавляют 210 мл метанола. Поддерживают при перемешивании в течение 16 часов в атмосфере азота при комнатной температуре. Концентрируют и получают 1,4 г продукта, который очищают хроматографией на двуокиси кремния, элюант 224 (93-7-0,4). Концентрируют и получают 0,305 г искомого сырого продукта, который перекристаллизовывают из простого изопропилового эфира, промывают, сушат при 50 при пониженном давлении. Таким образом получают 0,267 г искомого продукта с точкой плавления 222-231 . ЯМР 3 ппм.18(0,9 3). 0,843-2 1,01 -1,17 -1,243- 1,30 -1,38 , 1,34-1,47 6 и 12- 2,27 3,872 3,992 4,235 4,271 4,9413 7,265 7,52 7,20 в пара-положении 7,35 в мета-положении 7,76 в орто-положении. Приготовление 1 4-(4-фенил-1-имидазол-1-ил)бутанамин. Стадия 2-(4-(4-фенил-1-имидазол-1-ил)бутил)-1-изоиндол-1,3(2)дион. Прикапывают в течение 1 часа 30 минут раствор, содержащий 5,05 г 4-фенил-1-имидазола в 25 см 3 диметилформамида, в смесь 7 см 3 диметилформамида и 2,02 г гидрида натрия. Затем вводят 10,86 г -4 бромбутилфталимида, растворенного в 25 см 3 диметилформамида. Доводят полученный раствор до 70 в течение приблизительно 1 часа 30 минут. Оставляют до возвращения к комнатной температуре, концентрируют полученный раствор, поглощают водой, экстрагируют этилацетатом. Промывают органические фазы водой, сушат, фильтруют и концентрируют. Получают 15 г продукта, который перекристаллизовывают из этилацетата. Осушают полученный продукт, промывают его этилацетатом и сушат при пониженном давлении при 50 . Получают 5,5 г искомого продукта, с точкой плавления 130-132 . ЯМР 3 ппм. 1,75(2)-1,86(2) 2 центральные 3,742 4,03 2 7,222 4 7,261 3 7,36 2 3 и 5 7,565 около 7,734 около 7,862 и 6. Стадия 4-(4-фенил-1-имидазол-1-ил)бутанамин. Поддерживают при флегме в течение 8 часов смесь 3,45 г полученного на стадиипродукта, 100 мл этанола и 0,97 мл гидразингидрата. Концентрируют реакционную смесь, добавляют приблизительно 50 мл 2 н. гидроксида натрия, экстрагируют этилацетатом. Промывают органические фазы при помощи 2 н. гидроксида натрия, затем хлорида натрия. Сушат, фильтруют и концентрируют. Получают 2,21 г искомого продукта. ЯМР 3 ппм. 1,47- 1,872 центральные 2,73 , 3,97 -2-2 7,203 7,505 7,37 ( шир.) 2 3 и 5 7,241 4 7,772 2 и 6. Пример 2. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(3-имидазо(4,5-)пиридин-3-ил)бутил)иминоэритромицин. Растворяют 708,2 мг 2-ацетат-11-деокси-10,11-дидегидро-3-де 2,6-дидеокси-3 метил-3 метил-альфа-рибогексопиранозил)окси)-121-имидазол-1-ил)карбонил)-6 метил-3-оксо-эритромицина(полученного,как указано в примере 1 европейской заявки на патент 0596802) и 958 мг 3-имидазо-(4,5-)пиридин-3 бутанамина в 2,82 см 3 ацетонитрила и 0,28 см 3 воды. Доводят реакционную смесь до 80 . Оставляют до возвращения к комнатной температуре и выливают в раствор кислого фосфата натрия. Экстрагируют хлористым метиленом и промывают водой. Собирают водные фазы и снова экстрагируют. Сушат, фильтруют,споласкивают и получают 826 мг продукта. Растворяют полученный продукт в 16,5 см 3 метанола. Поддерживают реакционный раствор при перемешивании при комнатной температуре в течение 20 часов. Получают 789 мг искомого сырого продукта, который очищают хроматографией, элюант смесь хлористого метилена, метанола, раствора аммиака (94-16-0,4). Получают 327 мг искомого продукта с точкой плавления 200 .1313. ЯМР 3 400 мГц ппм. 12(полученного как указано в примере 1 европейской заявки на патент 0596802) в раствор, содержащий 953 мг 1-имидазо(4,5-)пиридин-1-бутанамина, 2,82 см 3 ацетонитрила и 0,28 см 3 воды. Доводят реакционную смесь до 55 . Поддерживают при этой температуре в течение 44 часов и добавляют 0,5 см 3 ацетонитрила. Продолжают нагревание при 55 в течение 20 часов. Оставляют до возвращения к комнатной температуре и выливают в насыщенный раствор кислого фосфата натрия. Экстрагируют водную фазу хлористым метиленом и промывают водой хлорметиленовую фазу. Сушат на сульфате натрия, фильтруют и выпаривают. Получают 806 мг продукта, к которому добавляют 16,1 см 3 метанола. Поддерживают реакционную смесь при комнатной температуре в течение 24 часов и выпаривают досуха. Получают 656 мг продукта, который хроматографируют на двуокиси кремния элюант смесь 223 (94-6-0,4). Получают сырой целевой продукт, который очищают хроматографией на силикагеле, элюируя смесью 34 (94-6-0,4). После растворения остатка в смеси этилацетат с простым изопропиловым эфиром, фильтрования и выпаривания досуха получают целевой продукт. Т. пл.203 .17,613. 0,813-2 1,00 -1,17 -1,25 -1,31 -1,383- 1,35 -1,476 и 12-3 1,68 и 1,93 центральные 2 цепи 2,27(3)2 2,616-3 2,453 около 2,60 ( в замаскированной части) 8 3,074 около 3,15 ( шир) 10 3,182 3,5611 3,535 3,60-3,80 2 3,872 около 4,2524,245 4,281 4,9113 7,21 (,5 и 8) 6 7,80 (,8 и 1,5) 7 ароматические 8,56 (,5 и 1,5) 5 8,15222. Приготовление 2 Получение аминов, используемых в качестве исходных продуктов в примерах 2 и 3 3-имидазо(4,5-)пиридин-3-бутанамин и 1-имидазо(4,5-)пиридин-1- бутанамин. СтадияВ раствор 5,95 г 4-азабензимидазола и 15,5 г -4-бромбутилфталимида в 30 см 3 диметилформамида добавляют 10,3 г карбоната калия. Смесь перемешивают 20 часов при комнатной температуре. Нерастворенный продукт фильтруют, споласкивают хлористым метиленом. Органическую фазу промывают водой, затем сушат на сульфате магния и выпаривают полученный маслянистый остаток промывают петролейным эфиром, затем изопропиловым эфиром. Получают 16,3 г сырого продукта, который очищают хроматографией на двуокиси кремния, элюируя смесью хлористого метилена с ацетоном, и получают 4,9 г продукта , т. пл.143 и 3,9 г продуктат. пл.172 . Стадия 1 3-имидазо(4,5-)пиридин-3-бутанамин (исходный продукт в примере 2). Доводят до температуры кипения в течение 19 часов смесь 32,86 г продукта , полученного выше, 697 см 3 этанола и 20 см 3 гидразина. Оставляют до возвращения к комнатной температуре. Фильтруют, споласкивают и выпаривают досуха. Поглощают хлористым метиленом, фильтруют, споласкивают и выпаривают досуха. Получают 18,87 г целевого продукта. ЯМР 3 - 250 мГц. 1,52 -2,002 2 центральные 1,63 ( - шир) подвижные 2 2,762-2-24,332 7,24 (,8 и 5) 6 8,08 (,8 и 1,5) 7 8,40 (,5 и 1,5) 5 8,082. Стадия 2 1-имидазо(4,5-)пиридин-1-бутанамин (исходный продукт в примере 3). Доводят до температуры кипения в течение 21 часа смесь 32 г продукта , полученного выше, 640 см 3 этанола и 24,8 см 3 гидразина. Оставляют до возвращения к комнатной температуре. Фильтруют, споласки 13 4746 1 вают этанолом и выпаривают при пониженном давлении. Поглощают хлористым метиленом, фильтруют,споласкивают и выпаривают досуха. Получают 19,5 г целевого продукта. ЯМР 3. 1,45 -1,962 центральные 2 2,742-2 около 1,45 подвижный 4,232 4746 1 Пример 15. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 5-(1-бензимидазол-1-ил)фенил)иминоэритромицин. Получен из 2-(4-бромпентил)-1-изоиндол-1,3-(2)-диона. Пример 16. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 5-хлор-1-бензимидазол-1-ил)бутил)имино)эритромицин. Т. пл.145-148 . Пример 17. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-12,11(оксикарбонил 4-(1-индол-1-ил)бутил)иминоэритромицин. Пример 18. 11,12-дидеокси-3-де(2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(1-метил-1-индол-4-ил)бутил)иминоэритромицин.20 ,13. Пример 19. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(2-фенил-4-хинолинил)бутил)иминоэритромицин. Т. пл.195-197 . Пример 20. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(1-бензотриазол-1-ил)бутил)иминоэритромицин. Т. пл.200-202 . Пример 21. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(2-бензотриазол-2-ил)бутил)иминоэритромицин. Т. пл.164-165 . Пример 22. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6-метил-3-оксо 12,11-(оксикарбонил 4-(5,6-диметил-1-бензимидазол-1-ил)бутил)иминоэритромицин. Т. пл.174-176 . Пример 23. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(3-хинолинил)бутил)иминоэритромицин. Т. пл.195-197 . Пример 24. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(2-хинолинил)бутил)иминоэритромицин. Т. пл.179-181 . Пример 25. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(2-метил-1-бензимидазол-1-ил)бутил)иминоэритромицин. Т.пл.128-132 . Пример 26. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(6-хлор-1-бензимидазол-1-ил)бутил)иминоэритромицин. Т. пл.192-194 . Пример 27. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(1-метил-1-бензимидазол-2-ил)бутил)иминоэритромицин. Пример 28. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(5-имидазо)-4,5-)пиридин-5-ил)бутил)иминоэритромицин.12,213. Пример 29. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)6 метил-3 оксо-12,11-(оксикарбонил 4-(4-(4-хлорфенил)-1-имидазол-1-ил)бутил)иминоэритромицин. Нагревают 7 часов до 751 г 2-ацетат-11-деокси-10,11-дидегидро-3-де 2,6-дидеокси-3 метил-3-метил-альфарибогексопиранозил)окси)-121-имидазол-1-ил)карбонил)6 метил-3-оксоэритромицина (полученного как указано в примере 1 европейской заявки на патент 0596802) в 4 см 3 4746 1 ацетонитрила, содержащего 10 воды с 1,4 г 4-(4-хлорфенил)-1-имидазол-1-бутанамина. Оставляют до возвращения к комнатной температуре, разбавляют водой, экстрагируют этилацетатом, сушат, выпаривают растворитель и получают 2,3 г продукта, ацетилированного в положении 2. Добавляют 60 мл метанола и поддерживают 16 часов при перемешивании, выпаривают растворитель, хроматографируют остаток на двуокиси кремния (элюент 224 95-5-0,4), концентрируют и кристаллизуют остаток из простого эфира. Сушат кристаллизованный продукт при пониженном давлении при 80 и получают 381 мг целевого продукта. Т. пл.192-194 . ЯМР 3 ппм. 0,833-2 1,00- 1,16 -1,24 -1,30- 1,383- 1,33- 1,476 и 122,26 3,872 4,235 4,281 4,932 Н 13 7,265 имидазола 7,502 имидазола 7,32-7,70 ароматические 3,51 . Приготовление 4-(4-хлорфенил)-1-имидазол-1-бутанамина, используемого в качестве исходного в примере 29. Стадия 4-(4-хлорфенил)-1-имидазол. Кипятят с обратным холодильником в течение 1 часа 23,34 г -бром-4-хлорацетофенона в 150 мл формамида оставляют охлаждаться, подщелачивают раствором гидроксида натрия, экстрагируют дихлорметаном, промывают водой, сушат, выпаривают растворитель, хроматографируют остаток на двуокиси кремния(элюент 224 8-2-0,04) и получают 13,4 г целевого продукта. Т. пл.146-148 . Стадия 2-(4-(4-(4-хлорфенил)-1-имидазол-1-ил)бутил)-1-изо-индол-1,3-(2)-дион. Работают как на стадииприготовления 1 примера 1, используя 12,2 г продукта, полученного на предыдущей стадии, 4,96 г гидрида натрия и 23,83 г -4-бромбутилфталимида. Получают 9,7 г целевого продукта. Стадия 4-(4-хлорфенил)-1-имидазол-1-бутанамин. Работают как на стадииприготовления 1 примера 1, но используя 14,2 г продукта, получаемого на стадии , и 3,6 мл гидразингидрата в 200 мл этанола. Получают 12 г сырого продукта, который хроматографируют на двуокиси кремния (элюент 22-4 8-2-0,04) и получают продукт, используемый как таковой для синтеза. ЯМР (3) ппм. 1,22 ( шир.) 2 подвижные 1,47 -1,88 центральные 2 2 2 2 23,982,74 2 7,19 (,1,5)-7,50 (,1,5) 2 и 5 7,33 и 7,70 ароматические. Пример 30. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(4-(2-метоксифенил)-1-имидазол-1-ил)бутил)иминоэритромицин. Нагревают при 80 в течение 8 часов 706 мг исходного соединения примера 29 в 3 мл ацетонитрила и 908 мг 4-(2-метоксифенил)-1-имидазол-1-бутанамина. Охлаждают до комнатной температуры, выливают в раствор гидрофосфата натрия (0,5 М), экстрагируют этилацетатом, промывают водой, сушат, выпаривают растворитель, получают 1,6 г продукта, ацетилированного в положении 2. Добавляют 50 мл метанола, перемешивают в течение 16 часов, выпаривают растворитель, хроматографируют остаток на двуокиси кремния(элюент - с 4 ) и кристаллизуют из простого эфира. Получают 194 мг целевого продукта. Т. пл.143-145 . ЯМР 3 ппм. 0,853-2 1,01 -1,16 -1,24 -1,30 -1,373- 1,34 -1,476 и 122,26 4746 1 Нагревают до температуры кипения 9,36 г 2-бром-2-метокси-ацетофенона в 50 мл формамида, оставляют до возвращения к комнатной температуре, промывают 2 н. раствором соляной кислоты, фильтруют, подщелачивают до 8-9 при помощи 2 н. гидроксида натрия, экстрагируют дихлорметаном, промывают водой,сушат, выпаривают растворитель, хроматографируют остаток на двуокиси кремния (элюент 22-4 95-5-0,4) и получают 6,15 г целевого продукта. Стадия 2-(4-(4-(2-метоксифенил)-1-имидазол-1-ил)бутил)-1-изоиндол-1,3(2)-дион. Работают как на стадииприготовления 1 примера 1, но используя 6 г продукта, полученного на стадии, 1,99 г гидрида натрия и 9,93 г -4-бромбутилфталимида. Получают 6,15 г целевого продукта. Стадия 4-(2-метоксифенил)-1-имидазол-1-бутанамин (фумарат). Работают как на стадииприготовления 1 примера 1, но используя 5,65 г продукта, полученного на стадии , и 1,45 мл гидразингидрата в 75 мл этанола. Получают 3,8 г сырого продукта, который растворяют в 4 мл тетрагидрофурана, затем добавляют 1,87 г фумаровой кислоты, растворенной в 20 мл метанола. Прибавляют 10 мл простого эфира, осушают образованные кристаллы, сушат их при 80 при пониженном давлении и рекуперируют 3,77 г фумарата целевого продукта. Т. пл.160-162 . ЯМР (3) ппм. 1,482-1,872 центральные 2 3,46 2 2,733 3,94 ф 3,97 2 6,946 7,04 -7,215 и 4 7,51 2 и 5 8,192. Пример 31. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(4-(4-фторфенил)-1-имидазол-1-ил)бутил)иминоэритромицин. Нагревают при 60 в течение 4 часов 30 минут 2,11 г исходного соединения примера 29 в 9 мл ацетонитрила и 2,8 г 4-(4-фторфенил)-1-имидазол-1-бутанамина. Охлаждают до комнатной температуры, выливают в воду, экстрагируют этилацетатом, промывают водой, сушат, выпаривают растворитель, получают 5,2 г продукта, ацетилированного в положении 2. Добавляют 20 мл метанола, перемешивают в течение 3 часов 30 минут, выпаривают растворитель, хроматографируют остаток на двуокиси кремния (элюент 22-4 95-5-0,3) и кристаллизуют из простого эфира. Получают 1,34 г целевого продукта. Т. пл.190-192 . ЯМР 3 ппм. 1,33 -1,476 и 122,272 2,616- 3,0 - 3,18 4 и 10 3,56113,98 2 3,59-3,81 2 около 7,05 - около 7,73 фторфенил 7,215 имидазола 7,492 имидазола. Приготовление 4-(4-фторфенил)-1-имидазол-1-бутанамина, используемого в качестве исходного в примере 31. Стадия 4-(4-фторфенил)-1-имидазол. Нагревают 2 часа при рефлюксе 10,85 г бромистого 4-фторфенацила в 60 мл формамида, оставляют до возвращения к комнатной температуре, подкисляют до 2 при помощи 1 н. соляной кислоты, фильтруют,нейтрализуют добавлением раствора аммиака, экстрагируют дихлорметаном, промывают водой, сушат, выпаривают растворитель, хроматографируют остаток на двуокиси кремния (элюент 224 95-5-0,4) и получают 5,8 г целевого продукта. Т. пл.130-132 . Стадия 2-(4-(4-(4-фторфенил)-1-имидазол-1-ил)бутил)-1-изоиндол-1,3(2)-дион. Работают как на стадииприготовления 1 примера 1, но используют 10 г продукта, полученного на стадии , 1,95 г гидрида натрия и 11,80 г -4-бромбутилфталимида. Получают 7,53 г целевого продукта. Т. пл.138-140 . Стадия 4-(4-фторфенил)-1-имидазол-1-бутанамин. Работают как на стадииприготовления 1 примера 1, но используют 3,64 г продукта, полученного выше на стадии , и 1 мл гидразингидрата в 80 мл этанола. Получают 2,4 г сырого продукта, который хроматографируют на двуокиси кремния (элюент 224 8-2-0,03) и получают продукт, используемый как таковой для синтеза. ЯМР (3) ппм. 1,48 -1,81 все центральные С 2 2,74-С 3 3,98-С 2-С 2 7,06 С 7,22 4746 1 Пример 32. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(7-метокси-4-хинолинил)бутил)иминоэритромицин. Нагревают при 50 в течение 53 часов 706 мг исходного соединения примера 29 в 4 мл ацетонитрила и 1,43 г 7-метоксихинолин-4-бутанамина. Оставляют до возвращения к комнатной температуре, выливают в раствор гидрофосфата натрия (0,5 М), экстрагируют дихлорметаном, промывают водой, сушат, выпаривают растворитель, получают 1,09 г продукта, ацетилированного в положении 2. Добавляют 10 мл метанола, перемешивают в течение 16 часов, выпаривают растворитель, хроматографируют остаток на двуокиси кремния (элюант 22- 95-5) и кристаллизуют из простого эфира. Получают 295 мг ожидаемого продукта. Т. пл.110 . ЯМР 3 ппм. 3,06-(2)2 - 3,702 3,95-3 7,12 -7,19 -7,42 -7,94 -8,70 пиридин. Приготовление 7-метоксихинолин-4-бутанамина, используемого в качестве исходного в примере 32. Стадиясоль трифенилфосфония -(3-бромпропил)фталимида. Нагревают до температуры кипения в течение 44 часов 13,4 г -бромпропилфталимида и 13,15 г трифенилфосфина, суспензированного в 75 мл ксилола. Оставляют до возвращения к комнатной температуре,осушают осадок, промывают его простым этиловым эфиром и сушат при пониженном давлении при 60 . Рекуперируют 24,88 г целевого продукта Т. пл.220-222 . Стадия-2-(4-(7-метокси-4-хинолинил)-3-бутенил)-1-изо-индол-1,3(2)-дион. Добавляют 4 г 7-метокси-4-хинолинилкарбоксальдегида в суспензию 12,47 г соли трифенилфосфония 3 бромпропилфталимида в 200 мл тетрагидрофурана. Охлаждают до -50 , добавляют 2,72 г трет-бутилата калия, оставляют до медленного повышения температуры до -6 , фильтруют, концентрируют фильтрат,поглощают остаток в этилацетате, промывают водой, сушат, выпаривают растворитель и рекуперируют 9,26 г сырого продукта, который хроматографируют на двуокиси кремния (элюент 3- 80-20, затем 7030). Рекуперируют 3,575 г целевого продукта. Стадия 2-(4-(7-метокси-4-хинолинил)бутил)-1-изоиндол-1,3(2)-дион. Растворяют 3,50 г продукта, полученного на стадии , в 50 мл метанола, добавляют 0,36 г палладия на активированном угле и гидрируют 3 часа при 600 мбар. Фильтруют, выпаривают растворитель и получают 3,48 г ожидаемого продукта. Стадия 7-метоксихинолинил-4-бутанамин. Растворяют в горячем виде 3,46 г продукта, полученного в стадии , в 70 мл этанола, добавляют 1,86 мл гидразингидрата, поддерживают при рефлюксе в течение 17 часов, удаляют фильтрованием осадок, выпаривают растворитель, поглощают остаток в 70 мл дихлорметана. Фильтруют, выпаривают растворитель и отбирают 2,19 г целевого продукта. ЯМР (3) ппм. 1,6 -1,79 центральные 2 2,75-2-(2)3 3,052-2 3,95-3 7,10 (,4,5)-7,21 -7,92 -8,71 (,4,5) хинолин. Пример 33. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(2-(2-пиридинил)-4-тиазолил)бутил)иминоэритромицин. Нагревают при 60 в течение 5 часов 705 мг исходного соединения примера 29 (полученного как указано в примере 1 европейской заявки на патент 0596802) в 3 мл ацетонитрила и 0,705 г 2-(2-пиридинил)тиазол)-4-бутанамина. Оставляют до возвращения к комнатной температуре, выливают в воду, экстрагируют этилацетатом, промывают водой, сушат, выпаривают растворитель, получают 1,8 г продукта, ацетилированного в положении 2. Добавляют 15 мл метанола, нагревают до температуры кипения в течение 2 часов, выпаривают растворитель, хроматографируют остаток на двуокиси кремния (элюент 224 95-5-0,3, затем - 9-1) и кристаллизуют из простого эфира. Получают 194 мг целевого продукта. Т. пл.157-159 . ЯМР (3) ппм. 1,33 и 1,47 6 и 122,26(3)2 4746 1 Стадия 2-аминокарбонилпиридин. Прикапывают 50 мл раствора диазометана (0,4 М/л) в раствор, содержащий 2 г пиколиновой кислоты, 20 мл дихлорметана и 5 мл метанола. После 30 минут перемешивания при комнатной температуре выпаривают растворитель при пониженном давлении, хроматографируют остаток на двуокиси кремния (петролейный эфир (60-80)- 5-5) и рекуперируют 1,48 г метилового эфира. Нагревают до 50 в течение 4 часов 1,42 г сложного эфира в 5 мл гидрата окиси аммония, оставляют до возвращения к комнатной температуре, экстрагируют простым эфиром, промывают водой, сушат, выпаривают растворитель и рекуперируют 1,05 г ожидаемого продукта. Т. пл.105 . Стадия 2-пиридинкарботиоамид. Медленно добавляют 43 г петнасульфида фосфора к 46,8 г амида, полученного на стадии , в 700 мл тетрагидрофурана. Перемешивают 4 часа при комнатной температуре, выливают в воду, экстрагируют простым эфиром, сушат и выпаривают растворитель при пониженном давлении. После хроматографии на двуокиси кремния (элюент 22- 8-2) извлекают 10 г ожидаемого продукта. Т. пл.137 . Стадия 2-(2-пиридинил)-4-тиазолэтилкарбоксилат. Прикапывают 16,3 мл этил-бромпирувата к 15,9 г продукта, полученного как на стадиив 250 мл этанола, и нагревают 5 часов при рефлюксе. Выпаривают растворитель при пониженном давлении, хроматографируют остаток на двуокиси кремния (элюент гексан- 1-1) и получают 10,2 г целевого продукта. Т. пл.69,1 . Стадия 2-(2-пиридинил)-4-тиазолметанол. Медленно добавляют 40 мл метанола в смесь, содержащую 9,3 г сложного эфира, полученного на стадии, и 4,1 г боргидрида натрия в 100 мл тетрагидрофурана и нагревают 2 часа при рефлюксе. Оставляют до возвращения к комнатной температуре, выливают в воду, нейтрализуют при помощи 1 н. соляной кислоты,экстрагируют дихлорметаном, сушат органическую фазу и выпаривают растворитель при пониженном давлении, хроматографируют остаток на двуокиси кремния 1 (элюент Е-22 1-1) и получают 5,8 г ожидаемого продукта. Т. пл.100 . Стадия 2-(2-пиридинил)-4-тиазолкарбоксальдегид. Поддерживают 2 часа при рефлюксе 5,8 г продукта, полученного на стадии , в 60 мл толуола в присутствии 13 г окиси марганца, фильтруют и выпаривают растворитель при пониженном давлении. Получают 5 г ожидаемого продукта. Т. пл.131 . Стадия-2-(4-(2-(2-пиридинил)тиазол)-3-бутенил)-1-изоиндол-1,3(2)-дион. Работают как на стадииприготовления примера 32, используя 5,70 г альдегида, полученного на стадии, и 15,9 г соли трифенилфосфония 3-бромпропилфталимида и 3,70 г трет-бутилата калия. Получают 8,73 г целевого продукта. Т. пл.139-141 . Стадия 2-(4-(2-(2-пиридинил)тиазол)бутил)-1-изоиндол-1,3(2)-дион. Работают как на стадииприготовления примера 32, используя в качестве исходного 7,22 г продукта,полученного на стадии , и 1,5 г палладия на активированном угле, гидрогенируя 2 часа при 1800 мбар. Получают 6,33 г целевого продукта. Т. пл.119-121 . Стадия 2-(2-пиридинил)тиазол-4-бутанамин. Работают как на стадииприготовления примера 32, используя 5,45 г продукта, полученного выше на предыдущей стадии, и 1,6 мл гидразингидрата, и нагревают 6 часов при флегме. Выпаривают растворитель,поглощают этилацетатом, промывают водой, сушат, выпаривают растворитель, хроматографируют остаток на двуокиси кремния (элюент 224 9-1-0,03) и получают 1,65 г ожидаемого продукта. ЯМР (3) ппм. 1,50 -1,82 центральные 2 2,76 -2,852-2-2 7,855 тиазола 7,315 7,784 8,183 8,616 1,402. Пример 34. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(4-(3-пиридинил)-1-имидазол-1-ил)бутил)иминоэритромицин. Нагревают до 70 в течение 20 часов 1 г исходного соединения примера 29 в 4 мл ацетонитрила и 936 мг 4-(4-(3-пиридинил)-1-имидазол-1-ил)бутанамина. Оставляют до возвращения к комнатной температуре,выливают в воду, экстрагируют этилацетатом, промывают водой, сушат, выпаривают растворитель, получают 1,34 г продукта, ацетилированного в положении 2. Добавляют туда 40 мл метанола, перемешивают в течение 2 часов, выпаривают растворитель, хроматографируют остаток на двуокиси кремния (элюент 22-4 95-5-0,4) и кристаллизуют из простого эфира. Получают 310 мг целевого продукта. Т. пл.187-188 . ЯМР (3) ппм. 2 2,453 2,626- 2,608 2,85-3,25 44 и 10, 2 3,525 3,5611 3,60 23,854,235 4,271 4,9313 7,293 пиридина 8,084 пиридинила 8,456 пиридина 8,972 пиридина 7,35 и 7,532 и 5 имидазола. Приготовление 4-(3-пиридинил)-1 имидазол-1-бутанамина, используемого в качестве исходного в примере 34. Стадия 2-(4-(3-пиридинил)-1-имидазол-1-ил)бутил-1-изоиндол-1,3(2)-дион. Работают как на стадииприготовления примера 1, используя 290 мг 3-пиридинил-1-имидазола, полученного как указано в . . . 753-5 (1938), 115 мг гидрида натрия и 633 мг -4-бромбутилфталимида. Получают 277 мг ожидаемого продукта. Т. пл.150-152 . Стадия 4-(3-пиридинил)-1-имидазол-1-бутанамин. Работают как на стадии В приготовления примера 1, используя 1,66 г продукта, полученного на стадии ,и 0,46 мл гидразингидрата в 30 мл этанола. Получают 936 мг продукта, используемого как таковой для синтеза. ЯМР (3) ппм. 1,49 -1,89 центральные 2 2,752-24,01 7,29 (,1)-7,55 (,1) 2 и 5 7,30 (в замаскированной части) 5 8,09 (,8 и 2) 4 8,47 (,5 и 2) 6 8,96 (,2) 2 1,49 ( шир.)2 подвижные. Пример 35. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(3-(3-пиридинил)-1-1,2,4-триазол-1-ил)бутил)иминоэритромицин. Нагревают до 75 в течение 8 часов 1 г исходного соединения примера 29 в 4 мл ацетонитрила и 1,21 г 4-(3-(3-пиридинил)-1-1,2,4-триазол-1-ил)бутанамина. Оставляют до возвращения к комнатной температуре,выливают в воду, экстрагируют этилацетатом, промывают водой, сушат, выпаривают растворитель, получают 2 г продукта, ацетилированного в положении 2. Добавляют 40 мл метанола, перемешивают в течение 16 часов, выпаривают растворитель, хроматографируют остаток на двуокиси кремния (элюент 22-4 90-10-0,04) и кристаллизуют из простого эфира. Получают 292 мг ожидаемого продукта. Т. пл.190-192 . ЯМР (3) ппм. 0,843-2 1,011,1681,2551,3041,3421,33 и 1,47 4 пиридина 8,626 пиридина 9,31 ( шир.) 2 пиридина. Приготовление 3-(3-пиридинил)-1-1,2,4-триазол-1-бутанамина, используемого в качестве исходного в примере 35. Стадия 2-(4-(3-(3-пиридинил)-1-1,2,4-триазол-1-ил)бутил-1- изоиндол-1,3(2)-дион. Работают как на стадииприготовления 1 примера 1, используя 2,1 г 3-пиридинил-1-1,2,4-триазола,полученного как указано в . . . (44)33, 4160-4164 (1979), 1,02 г гидрида натрия и 4,13 г -4 бромбутилфталимида. Получают 2,4 г целевого продукта. Т. пл.150-152 . Стадия 3-(3-пиридинил)-1-1,2,4-триазол-1-бутанамин (фумарат). Работают как на стадииприготовления 1 примера 1, используя 3,46 г продукта, полученного на стадии 4746 1 Работают как указано выше, но используя соответствующие амины, получают следующие продукты Пример 36. 11,12-дидеокси-3-де(2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(3-хинолинил)бутил)иминоэритромицин. Т. пл.190-192 . Пример 37. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(4-(4-метоксифенил)-1-имидазол-1-ил)бутил)иминоэритромицин. Т. пл.152-154 . Пример 38. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(2-фенил-4-тиазолил)бутил)иминоэритромицин. Т. пл.141-143 . Пример 39. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(4-(3-метоксифенил)-1-имидазол-1-ил)бутил)аминоэритромицин. Т. пл.144-146 . Пример 40. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(4,5-дифенил)-1-имидазол-1-ил)бутил)иминоэритромицин. Т. пл.180-182 . Пример 41. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(4-хиназолинил)бутил)иминоэритромицин. Т. пл.212-214 . Пример 42. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(2-(4-пиридинил)-4-тиазолил)бутил)иминоэритромицин. Т. пл.192-194 . Пример 43. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(1,2,3,6-тетрагидро-1,3-диметил-2,6-диоксо-7-пурин-7-ил)бутил)иминоэритромицин. Т. пл.251-253 . Пример 44. 11,12-дидеокси-3-де 2,6-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо-12,11(оксикарбонил 4-(4-(4-трифторметокси)фенил)-1-имидазол-4-ил)бутил)иминоэритромицин. Т. пл.168-170 . Амины, используемые как исходные продукты, получают следующими методами- Когда цепь связана с углеродом, например,(2)42 можно исходить из соответствующих альдегидов Амины, используемые для получения продуктов примеров 4, 8, 11, 12, 18, 19, 23 и 24, получали этим способом.- Когда цепь связана с азотом, можно получать амиды следующим способом Амины, используемые для получения продуктов примеров 1, 2, 3, 5, 9, 13 - 17, 20, 21, 22, 25, 26 и 28 получали этим способом.- Некоторые амины получают особым способом образуют гетероцикл и одновременно вводят цепь(примеры 6, 7, 10 и 27). Примеры фармацевтических составов Получали соединения, содержащие Продукт примера 1 - 150 мг эксципиент, в достаточном количестве - 1 г части эксципиента крахмал,тальк, стеарат магния. Продукт примера 2 - 150 мг эксципиент, в достаточном количестве - 1 г части эксципиента крахмал,тальк, стеарат магния. Продукт примера 3 - 150 мг эксципиент, в достаточном количестве - 1 г части эксципиента крахмал,тальк, стеарат магния. Фармакологическое исследование продуктов по изобретению Метод растворения в жидкой среде. Приготовляют серию пробирок, в которые помещают одинаковое количество стерильной питательной среды. Распределяют в каждую пробирку возрастающие количества исследуемого продукта, затем каждую пробирку засевают бактерийным штаммом. После инкубации в течение 24 часов в сушильном шкафу при 37 оценивают торможение роста просвечиванием, которое позволяет определять минимальные концентрации торможения , выраженные в микрограмм/см 3. 22 4746 1 Были получены следующие результаты (см. таблицу). Кроме того, продукты примеров 1, 2 и 3 показали интересную активность на бактерийных источниках относительно следующих 3513, 35112, 3511 и 3516. Штаммы грамположительных бактерий Штаммы грамположительных бактерий Соединениее 023102050210216 В 02110305 Национальный центр интеллектуальной собственности. 220072, г. Минск, проспект Ф. Скорины, 66. 23
МПК / Метки
МПК: C07D 417/04, C07D 471/04, C07D 401/04, C07H 17/08, A61P 31/04, C07D 215/02, A61K 31/7048
Метки: композиция, промежуточные, способ, основе, фармацевтическая, производные, эритромицина, соединения, получения
Код ссылки
<a href="https://by.patents.su/23-4746-proizvodnye-eritromicina-sposob-ih-polucheniya-farmacevticheskaya-kompoziciya-na-ih-osnove-i-promezhutochnye-soedineniya.html" rel="bookmark" title="База патентов Беларуси">Производные эритромицина, способ их получения, фармацевтическая композиция на их основе и промежуточные соединения</a>
Производные 5,6-дигидропирона и их фармацевтически приемлемые соли, промежуточные соединения для их получения, фармацевтическая композиция с антивирусной и антибактериальной активностью, способ лечения вызванных ретровирусом инфекции или заболевания

Номер патента: 4282
Опубликовано: 30.03.2002
Авторы: Элизабет ЛАННЕЙ, Брадлей Дин ТЕЙТ, Эдмунд Ли ЭЛЛСВОРТ
МПК: A61K 31/351, C07D 309/32, A61P 31/12...
Метки: соли, заболевания, композиция, ретровирусом, или, фармацевтически, приемлемые, вызванных, соединения, инфекции, промежуточные, производные, фармацевтическая, способ, антивирусной, получения, активностью, лечения, 5,6-дигидропирона, антибактериальной
Текст:
...кислоты. 1. Производные 5,6-дигидропирона по п. 8, где К 3 - группа формулы -Аг 2-СН 23 П 1 п 4-К 12. Производные 5,6-дигидропирона по п. 11, выбранные из группы, включающей 56-дигидро-4 окси-б-(Зметилбутил)-3-2(1-метилэтил)фенилтио 6-фенил 2 Н-пиран-2-он,3-5-этил-2-(1-метил-2-оксиэтил)фенилтио 5,6-ди 1 идро-4 окси-6,6-дифенил-2 Н-пиран-2-он 5-5-(2-циклопентил-5-изопропилфенил)тио-3,6-дигидро 4 окси 6 оксо 2 фенил-2 Нпиран-2-илпентановую...
Производные эритромицина, способ их получения и фармацевтическая композиция на их основе
Номер патента: 4361
Опубликовано: 30.03.2002
Авторы: Константин АГУРИДАС, Одиль ЛЕ МАРТРЕ, Янник БЕНЕДЕТТИ, Жан-Франсуа ШАНТО, Алексис ДЕНИС
МПК: C07H 17/08, A61K 31/70, A61P 31/04...
Метки: получения, эритромицина, композиция, производные, основе, способ, фармацевтическая
Текст:
...соли калия, которую растворяют в 1,2 л воды, подкисляют до рН 1 раствором концентрированной соляной кислоты после фильтрования, получают 29 г целевого продукта рассчитанный 58,52 3,43 6,82 15,6 обнаруженный 58,5 3,7 6,8 15,2. Стадия С 2-фенил-5-тиазолкарбоксилатметил. К раствору 4,77 г кислоты, полученной на стадии В, в 160 мл метанола, прибавляют 2,5 мл ацетилхлорида и осуществляют нагрев при рефлюксе в течение 18 ч. При пониженном...
Производные N-(4-пиперидинил) (дигидробензофуран или дигидро-2H-бензопиран)карбоксиамида, способ их получения, фармацевтическая композиция на их основе, промежуточные соединения

Номер патента: 4186
Опубликовано: 30.12.2001
Авторы: БОСМАНС, Ян-Паул Рене Мари Андре, ДЕ КЛЕЙН, Михел Анна Йозеф, ВАН ДАЛЕ, Георгес Хенри Паул
МПК: A61K 31/4523, A61K 31/16, A61P 1/14...
Метки: основе, способ, соединения, композиция, дигидро-2h-бензопиран)карбоксиамида, производные, фармацевтическая, или, получения, дигидробензофуран, промежуточные, n-(4-пиперидинил
Текст:
...гидроксидов, адкоксидов, гидридов, амидов или оксидов (карбонат натрия, гидрокарбонат натрия, карбонат калия, гидроксид натрия, метоксид натрия, гидрид натрия, амид натрия, карбонат кальция, гидроксид кальция, оксид кальция и др.), или неорганические основания, например третичный амин (,-диэтилэтанамин, -/1-метилэтил/-2-пропанамин, 4 этилморфолин и др.), чтобы нужная кислота нейтрализовалась в процессе реакции. В некоторых случаях...
Алкильные производные тразодона, способ их получения, промежуточные соединения и фармацевтическая композиция

Номер патента: 3830
Опубликовано: 30.03.2001
Автор: БАЙОККИ Леандро
МПК: C07C 229/16, A61K 31/437, A61P 25/24...
Метки: соединения, тразодона, способ, фармацевтическая, промежуточные, композиция, алкильные, производные, получения
Текст:
...к обычным лекарственным средам эти композиции могут содержать соответствующие фармацевтические добавки, такие как консерванты, стабилизаторы, эмульгаторы, соли для регулировки осмотического давления, буферы, ароматизирующие и окрашивающие компоненты. При специальных терапевтических предписаниях композиции в соответствии с настоящим изобретение могут включать другие совместимые активные ингредиенты, сопутствующее назначение которых полезно...
Соединения, селективно ингибирующие ароматазу, способ их получения, фармацевтическая композиция на их основе
Номер патента: 4357
Опубликовано: 30.03.2002
Авторы: РИСТО АРВО САКАРИ ЛАММИНТАУСТА, Рейно Олави ПЕЛЬКОНЕН, Ярмо Сакари САЛОНЕН, Марья-Лииса СЕДЕРВАЛЛ, Айре Марья ЛАЙНЕ, Арья Маркетта КАЛАПУДАС, Арто Йоханнес КАРЬЯЛАЙНЕН
МПК: A61K 31/41, A61K 31/425, A61P 43/00...
Метки: соединения, способ, получения, композиция, основе, ароматазу, фармацевтическая, ингибирующие, селективно
Текст:
...опухолей молочной железы синтезируют эстрадиол и эстрони поэтому происходит непрерывное стимулирование роста опухоли(.,59 770-782, 1987). Способность соединений изобретения ингибировать фермент ароматазу показали способом испытания, описанным М(, . 6,2, 1965, . 94-99). Применяли ароматазу человека. Фермент получали из плаценты человека, которая богата этим ферментом. Микросомную фракцию (1000000 х г осадка) получали центрифугированием....
Предыдущий патент: Способ получения ферритина
Следующий патент: Замещенные ароматические амиды тиокарбоновой кислоты и гербицидное средство, содержащее их
Случайный патент: Способ формирования цифрового радиоканала от подвижного объекта к неподвижному пункту управления