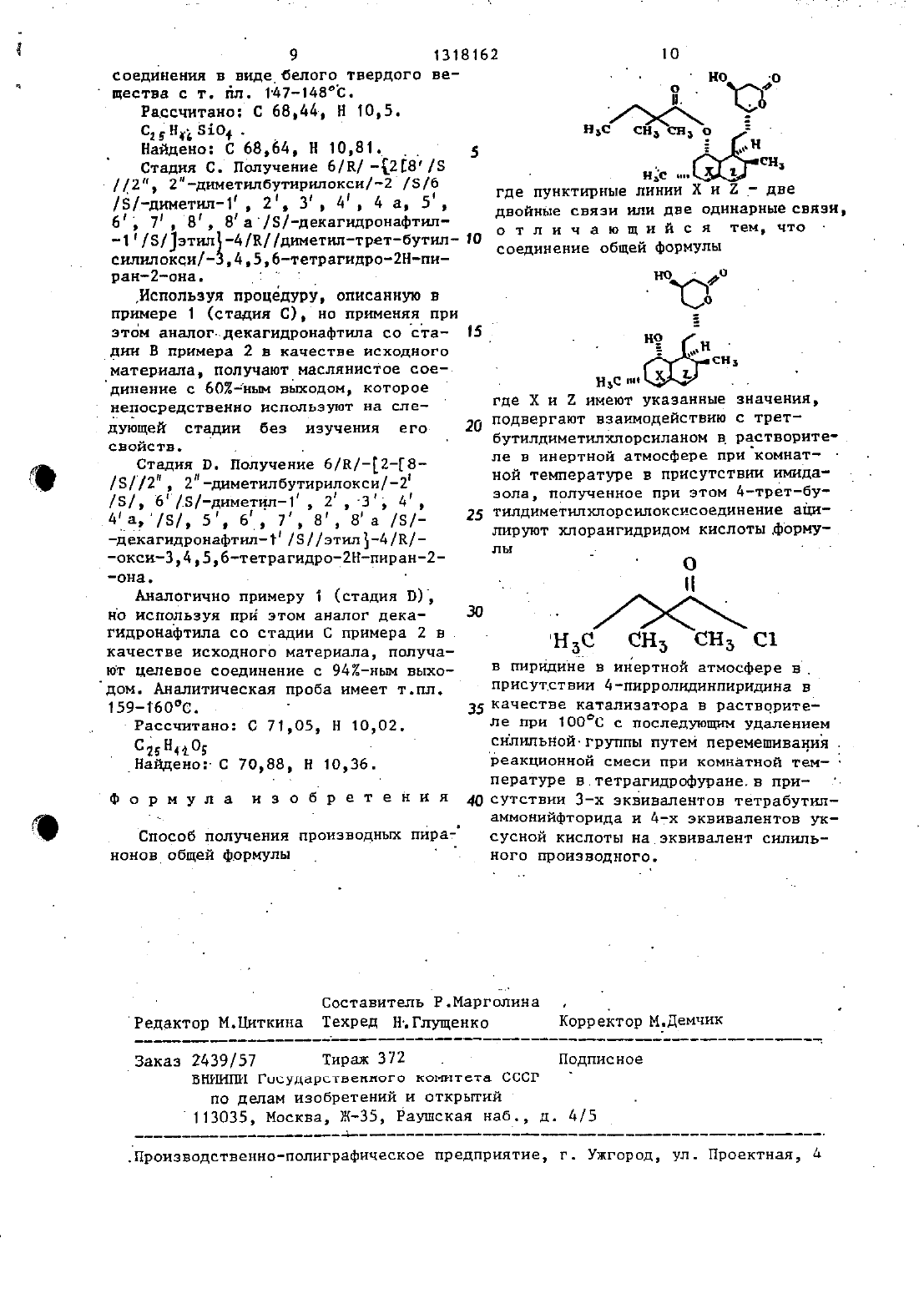
Способ получения производных пиранонов
Номер патента: 504
Опубликовано: 30.03.1995
Авторы: Элвин К. Виллард, Роберт Л. Смит, Вильям Ф. Хоффман





Текст
Изобретение относится к способу получения новых производных пиранонов общей формулы 1НЪС у р где пунктнрные линии Х и 2 представляют две двойные связи или две одннарные связи, проявляющих антигипер холестеролемическую активностьд- Целью изобретения является получение соединений, обладающих более высокой активностью.вано в качестве антигиперхолесте ролемического агента для печения атеч росклероза гиперлипемии и других пободныи заболеваний человека. Его можно применять стоматическим или парентеральным способом в виде капсул,таблетокпрепаратов для инъекций.кий способ. Дозы могут варьироваться а зависимости от возраста пациента,его состояния, веса и других показателей. Дневная Доза для взрослого пациеита изменяется от 2 до 2000 мг(предпочтительно от 10 10 О мг) и может быть разделена на три или четыре приема. Можно применять более высокие дозы. Как соединение 11 формулыизвестное под названием мевинолин,так И предлагаемое СОЕДИНЕНИЕ ПРОЯВЛНЮТ антигиперхолестеролемическую активность. Однако последний более активен, показателем чего является ферментно ингибирующая активностьСоединение 1, получаемое при помо . нового способа, может быть испольоСоединения были исследованы В форме соли натрия разомкнутой оксикис лоты. Они были приготовлены в видерастворов Д мг/млв 102 этанола. Перед проведением анализа препарации разбавляли в ДМСО-диметилсульфоксиде. НС определяли с использованием пяти концентраций для каждого ингибитора в анализе НМС СоАредуктазы. Изоляция и анализ на НМСтСоАредукМикросомы получали из печени крыс,которым добавляли в корм холестирамн в течение 7 дней. НМССоАредуктазу подвергали сольюбипиэацин из микросом методом Геллера и Шрюсбери и затем очистке через стадию осаждения вторичньпдсульфатом амония. Дрепарат фермента хранили при температуре 8 ОС в виде 100 Н л проб и она оставалась стабильной в течение по край Ней мере 3 мес. Перед использованиемфермент активировали при 37 С в тече 353,5 ММ ЭЦТК, рН 7,0, 10 ММ дитиотрей топа, альбумин бычьей сыворотки с концентрацией 0,1 мг/мл, 0,О 2 ь Кн ЕСНМС СоА с указанной концентрацией, 0,3 Н г частично очищенного фермента (удельная активность 1 ОО 150 имольмин мг ) с ИНГНбИТОРОМг Спустя 5 мин инкубированин.с ингибитором и НАДПГ при 37 С реакцию инициировали при помощи 0,2 мМ НАДПГ(1 Д 5 м,5 др.ЮЦнмщш).Ршшщш прерывали при помощи 20 р л 5 М раствора НС 1. После Дополнительного инч кубирования в течение 15 мн при З 7 С для тото чтобы дать возможность закончить полную лактониэацию продукта,смесьпропускали через колонну 05 х х 5 см, содержащую БиоРекс в виде лорошка 100-200 меш (диаметр частиц 0,1 д 90,07 д мм) в хлоридной форме,который уравновешивали дистиллирован ной водой. При помощ этой смолы непрореагировавшй С НМС-СОА адсорбировался а продукт злюировали при помощи 3 мл дистиллированной воды непосредственно в ампулы для ана-. лиза сцинтилляций. После добавления 10 мл Аквазола 11 измеряли радиоактивность проб при помощи счетчикасцинтиллнций типа Пакард Б 2 д 50. Испытуемые ингибиторы превращали в соли натрия.Острое ингибирование синтеза холестерина у крыс. 5 Самцы белых крыс Гольцмана имелиих выдерживали на дисс те Пурина Формулэб-5008 в течение 1 недели. На седьмой день крыс раздели- . ли на семь или девять групп по 10 жи-10 вотныи с одинаковьш средним весом(приблизительно 160 г) в группе,кормление на основе диеты Формулзб продолжали. По 10 животных помещали в клетки, дно которык было изготовлено 15 из проволоки, клетки помещали в комнату с кондициоиированны воздухом.В 8 ч 30 мин на следующий день корм удаляли из клеток и каждой группекрыс давали через трубку, введенную до в желудок одну дозу испытываемого 7-соединения, суспендированного в 52 ном Эмульфоре в соляном растворе. Контрольная группа получала только суспендирующий носитель. СПУСТЯ 1 Ч 25 после введения соединения крысам внутрибрюшинным способом вводили 1-1 Сацетат натрия (2 б 7 р Ки/мл,удельная активность 231 р Кн/мг) вд мл крови через иглу, вставленнуюВ сердце, при этом животные предварительно подвергались легкой анестезии при помощи пентобарбитала затем попученные пробы помещали в пробирки, 35 содержащие 02 мл 0,4 М раствора цит рата натрия. Плазму, полученную при центрифугировании подвергали гидролизации а ХОЛЕСТЕРИН ЭКСТРЗГИРОВЗЛИдв петролейном эфире. Для измерения 40 содержания С холестерина в плаз ме 2 мл раствора в петролейном эфире выпаривали до сухого состояния в 8 ммллнметровы ампулах с делениямии содержание С ОПРЕДЕПЯПИ С НС пользованием жидкостного счетчикас сцинтилляций типа ПРИЗС ПЛд 50 Исследование на собаках.Чистопороднык самцов гончих помещали отдельно ДРУГ от друга и кормш пи измельченньм кормом на основе диеты Пурина Пзб Кейнин в расчете 30 г/кг 55 в день. Перед началом эксперимента у собак брали кровь дважды в неделю из шейной вены и определяли содержание холестерина плазмы до тек пор, Пока Полученные значения не станут стабильньми. Для того чтобы опреде лить воздействие мевинолина и соединения 1 на холестерин плазмы, собакам давали соединение в расчетеВ МГ/КГ В день. Его подмешмвалн в корм собакам. При этом продолжали брать пробы крови два раза в неделю еще в течение 4 недель и определяли содержание холестерина плазмы.0 СТРое ингибирование синтеза коз лестерина у крыс.Не было возможности установить гипохолестеролемическую активность у крыс при использовании МЛ 2 З 6 Б иначед как только после обработки препаратом ТРИТОН ИК-1334. Однако после стоматне ческого введения одной дозы испытуе мык ингибиторов синтез холестерина изС 3 ацетата у нормальных крыс зач метно иигибировалсн на 28 ч после обработки. Предварительные эксперименты как с мевинолинатом натрин,так и с соединениями 1 показали, что ингибиторная активность проявлялась у крыс очень быстро.-Спустя 1 ч после применения соединений внутрибрюшинньш способом вводили С ацетат(80 у Кн/кг), а спустя 50 мин опредеПЯПИ С 0 деРжаниеС 3 холестерина плазмы, КОТОРОЕ И служло мерой синтеза холестерина. Соль натрия соединений Т бЬШа Приблизительно в 2 раза более ЗКТИВНЗ ПО СРЭБНЕНИЮ С СОЛЬЮ НЗТРНЯ мевинолнната причем дозы с 50-ны ингибированием составили 0,046.(1978 ммоль) гидрата гидроокиси лит тия в 600 мл воды перемешивают при температуре кипения с обратньш холодильником в атмосфере азота в течение 56 ч. Реакционную смесь стаждают до 0 С а затем обрабатываютванной соляной кислоты. Далее эту смесь подвергают трехкратной зкстт ракционной обработке 250 миллиметровыми порциями диэтилового эфира и объединенные экстракты последовательно промывают тремя 200-мнллилитровыи порцями воды и затем 200 млиасьщеиного рассола. После сушки над сульфа- 5 том магния этот органический растворпр 0 фильтровывают и растворитель вы паривают в вакууме с получением маслоподобиого остатка. Остаток растворяют в 200 мл толуола и выдерживают 10 при температуре кипения с обратным холодильником В атмосфере азота в течение 2 чпри непрерывном отделении воды с целью обеспечить повторную лактонизацию. ния толуола н растирания остатка в гексане получают 5,15 г (812) указанного соединенин 1 Чд (К-метил) в виде белого твердого продукта, кото рый не требует дальнейшей очистки. 20Аналитическую пробу получают перекристаллизацией части этого продук та ИЗ УТИЛХЛОРИДЗ, В РЕЗУПЬТЕТЕ че го образуются белые сгустки с т.пл. 128-131 С Св вакууме). 25Смесь спирта со стадии А (183 г, 571 мысль), 21,5 г (1 д 28 мысль) трет-бутилдиметилхлорсилана и 19,4 г 45(285,6 ммоль) имидазола в 200 мл ЫЫ диметилформамида перемешивают при 7 20 С в атмосфере азота в-течение 18 чд Затем реакционную смесь разбавляют при помощи 1500 мл простого эфира и промывают последовательно водой, 22 НЫМ ВОДНЬЩ РЗСТВОРОМ ХЛОРИСТОВОДОРОДной кислоты, водой и насыщеннм раствором бикарбоната натрия. Эфирный раствор сушат Над сульфатом магния,фильтруют и выпаривают до объема 1 л. После Добавления 600 мл гекса-1 на объем уменьшают до 600 мл в паровой бане. Продукт кристаллизуют прикомнатной температуре, после изоляции и сушки воздухом получают 13,7 белого хлопкообразного твердого вещества. маточный раствор упарнвают до объема 250 мл и дополнительные кристаллы изолируют после того, как раствор вьщерживают при температуре 0 С в течение ночи. Общий вьшод 17,13 г (691) целевого соединения в виде белого хлопкообразного твердого вещества, температура плавления 142144 С (вакуум). ям (спс 1,) 0,10 (с, 6(СН 3)251) 0,90 (с, 9, (сн)с 51) 1,19 (д. 3,л 7 Гц, сна) 2,58 (д,2 д 4 ГцС 3 Н пнрана) д,3 (м , 2, сдн пирана и СБН нафталина) 4,70 (м, 1, сьн пирана)5,57 (м, 1, сдн нафталина) 5,58 (дд, 1, 3 6,10 Гц, Сдн нафталинз) 6,03 (д, 1, 3 10 гц, с,н нафталина). Рассчитано С 15 Н 1 О 451 Найдено С 69,46, Н 9,83. . Стадия С. Получение 6/К-2-/8/5/-2 2-диметилбутирилокси 2/З/6/К/ с 69,08, н 9,74.(20 ммоль) добавляют в перемешиваемьй магнитной мешалкой раствор 2,17 г(5 ммоль) спирта со стадии В и 74 мг 4 пирролидинпирицииа в 25 мл пиридина. Эту реакционную смесь перемешивают прн 100 С в атмосфере азота Ач Реакционную смесь разбавляют 250 мл простого эфира и промывают.1 Ы раствором НС 1 до тен пор, пока промывочная жидкость Не станет кислотной,а затем соляным раствором (3 х 5 О мл). После сушки над сульфатом магния раствор фильтруют и выпаривают, в результате чего получают 3,9 Г оранжевого масла. Это масло подвергают хроматографии в колонне 6 х 15 см на сипикагеле (23 ОЬОО меш или с размером частиц 0,0 б 80,037 мм). В результа 50 те элюировании (при атмосферном дав лении) смесью простой зфиргексаН(11 по объему) получают 2,7 г сое динения в виде вязкого желтого масла. ямт ссрс 1,)Ы 0,08 (с, 6, (сна), 55 5) 0,9 (5, 9, (сн,)дсз 1)тт 2 (с, 6, (сн 3)2 с) 2,50 (д, 2, 3 с 4 гц СЭН ПИРаНВ)1 4.33 (М, 1, С.Н пирана) д 63 (М) 19 Сан ПИраНа 3 543 (М,-4/К/окси-3,4,56 тетрагидро 2 Нпи ран-2-она. Раствор 2,7 г (5 ммолъ) сипилового простого эфира, 1,2 г (20 ммоль) уксусной кислоты и д,7 г (15 мысль) ВгдНГ-ЗНЯО в 25 мл ТГФ перемешвают при окружающей температуре в течение 18 ч. Реакционный раствор разбавляют 300 мл простого эфира И промывают последовательно 2-ным водным раствором НС 1 и насыщенным солнным раствором. После сушки (сульфат магния) и фильтрации раствор выаривают с тем, чтобы получить 2,8 г оранжевого масла. Это масло подвергают хроматографии на колонне 6 х 15 см с сн ликагелем (230-400 меш или с размером частиц 00680,О 37 м). В результате элюировании при атмосферном давлении смесью ацетон-метилен хлорид (119 по объему) получали 1 г(482) целевого соединения в виде бес Йцветного ТВЕРДОГО вещества, КОТОРОЕ.подвергают кристаллизации из нВхС 1 гексане, В РЗЗУЛЬТЗТЕ ЧЕГО ПОЛУЧАЮТ бесцветное твердое вещество с т.пл.8 декагидронафтнл 1/5//этилт 4/В/-оксит 3 д,5,бстетрагндро 2 Нпиран 2 она. Раствор 2,0 г (6,2 ммоль) спирта из примера 1 (стадия А) в 100 мл этилацетата подвергают гидрогениза ции в присутствии окиси платины (1 г) при давлении 2,813 атм до тех пор,пока не будет зафиксировано вьщеле ние 2 мольтэкв водорода. Кристаллы Удаляют фипьтрацей, а фильтрат выпарнвают до сухого состояния в результате чего получают белое твердое вещество (19 г), которое подвергают хроматографии на колонне 6 х 20 см ссипикагелем (230-400 меш или с размером частиц 0068 О,0 З 7 мм). В резуль тате элюировании при атмосферном(37 по объему получают 1,0 Ь (502) целевото соединения в виде бесцветного твердого вещества. А Аналитические пробы получают при помощи рекристаллизации порции этого материала изхлороформа, в результа те чего получают белое хлопкообраэ ное твердое вещество с т. пл. 166168 С. 7 Рассчитано С 70,33, Н с 1 вН 204 Найдено С 69,97, Н 10,05./5/декагидронафтип-1/5/зтип 4/5//диметнлтретбутилсилилокси/3,4,5,6 тетрагидро 2 Нпиран 2 она. Раствор спирта со стадии А (1,0 г,3,1 г ммоль), имидаэола (1,05 г,15,4 моль) н третбутилдиметилхлор силана (1,16 г, 7,7 моль) в 20 мл ЫЫ-диметилформамида перемешивают при 20 С в атмосфере азота в течениепри помощи 200 мл простого эфира ипромывают последовательно водой,22 ным водньм раствором хлористовочдородной кислоты и соляным раствором, Эфирный раствор сушат Над СУПЪФВТОМ магнии и вьшаривают, в результате чего получают белое твердое веществографин на колонне 6 х 2 О см с двуокисьюкремния (23 Од 00 меш или с размером частиц 0,068-0,037 мм). В результате элюировании при атмосферном давлении
МПК / Метки
МПК: A61K 31/365, C07D 309/30
Метки: получения, способ, производных, пиранонов
Код ссылки
<a href="https://by.patents.su/6-504-sposob-polucheniya-proizvodnyh-piranonov.html" rel="bookmark" title="База патентов Беларуси">Способ получения производных пиранонов</a>
Способ получения производных дипептидов

Номер патента: 503
Опубликовано: 30.03.1995
Авторы: Эдвард В. Тристрэм, Артур А. Пэтчетт, Элберт И. Тристрэм, Мэтью Дж. Виврэтт
МПК: A61K 37/02, C07K 5/06
Метки: дипептидов, способ, получения, производных
Текст:
...через колонку с сильнокислотной ионообменной смолой. Целевой продукт эюируют с помощью 2 ного раствора пиридина в воде. В итоге получают 0,36 г сырого продукта, который дополнителъио очищают с помощью колоночной хроматографии У полистирольной смоле ХАЛ-2.(меш. 40- -400), используя в качестве злента 0,1 М раствор гидроокиси аммония н смеси вода - метанол, 955(объем). Первьй изомер выиодитиз колонки после пропусканин 375-дООмл 45 элюеита, а...
Способ получения сульфинильных производных гетероциклических соединений или их фармацевтически приемлемых солей

Номер патента: 77
Опубликовано: 30.09.1994
Авторы: Дэвид Кокс, Джон Льюис Сучитский, Энтони Говард Инголл
МПК: A61K 31/415, C07D 235/28, C07D 263/58...
Метки: гетероциклических, солей, способ, или, производных, фармацевтически, соединений, получения, приемлемых, сульфинильных
Текст:
...25 меру 1 а. 7 - 3(1 Н 2 Бензимидазолипсульфиннл метил)-ЫПдиметил 2 пиридинамин (е)г Продукт ц превращают в целевое соединение (т.пл. 12 д 126 С)по прн- за меру 1 Ь П р им е р 5. 2-(Н 2 Бензиидазолилсульфинилметил)дбенволамнн. Ы 2 дХлорметнлфенил 32 д,6 триметилбенволсульфонамнд (а). Ы(2 Гидроксиметилфенил)-24,6-триметилбенэопсулЬфонамид(40 Г) в сухом дихлорэтана (80 мл) обрабатывают тионипхлоридом 01,15 ил) ПРИ КОМ натной температуре...
Способ получения производных стильбена

Номер патента: 258
Опубликовано: 30.12.1994
Авторы: Эккехард Вейс, Михаэл Клаус, Петер Мор
МПК: C07D 295/084, C07D 295/13, A61K 31/535...
Метки: получения, стильбена, производных, способ
Текст:
...179 г 1-2-9-(1 Е/-2-/56.7.8-тетрагидро-5.5.8.8 тетраметил-2-нафтил/ пропенищфеноксиэтилпиперидина в виде белых кристаллов. Тпл 91-93 С П р и м е р 4. Аналогично примеру 1 из 30,6 г бромида 2-(5.б.7.8-тетрагидро-5.5.8.8 тетраметил-2-нафтил)этил трифенилфосфония и 12.5 г 4-(2-морфолиноэтилтифбензальдегида получают 13.9 г 42-(р-/Е/-2-/5.6,7,8-тетрагидро-5.5.8 В-тетраметил-Ънафтип/ пропенил)фениптиоэтилморфолина. Тдд 125-127 С.П р и м е р...
Способ получения производных 5/6/-тиобензимидазола

Номер патента: 350
Опубликовано: 30.12.1994
Авторы: Дьердь Кермеци, Чаба Генци, Деже Корбонитш, Габорне Чер, Эндре Палоши, Гергей Хейа, Пал Кишш, Дьердьнь Свобода, Тиборне Сомор, Андраш Келемен
МПК: C07D 235/30
Метки: способ, получения, производных
Текст:
...В результате получают 5(6)-аллилтиобензимидаэолил-2-метил-карбамат 56)-пропин 2-илтио 1 бена имидазолил-2-метилкарбамат 5(6)-(бензилтио)бензимидазолил-2-метилкарбамат 5(6)-(д-нитрофенилтио)бензимидазолил-2-метипкарбамат 516)-(2 Ц-динитрофенилтио)бензимидазолил-2-метилкарбамат. 1П р И м е р д.Готовят суспензию 0,0 г 2-(метоксикарбониламино)бензимидазол-5(6)-илдисульфида в 30 мл метанола и добавляют к ней раствор 1,12 г гидроокиси калия в...
Способ получения производных фенилпиридазина

Номер патента: 497
Опубликовано: 30.03.1995
Авторы: Франц Ранингер, Энгельберт Клоймштайн
МПК: C07D 237/12, C07D 237/14, A01N 43/58...
Метки: получения, производных, способ, фенилпиридазина
Текст:
...легкую по ц. ью 44,7-ного водного раствора гид- удельному весу фазу освобождают оксида калия устанавливают рН 9,5. до, от растворителя в вакууме на рота- д Образуется прозрачный раствор и рас ционном испарителе и получают ходуется 17 д,д г раствора гидрок- 146,0 г маслянистогоО 3 феиилсида калия. Этот раствор охлаж- 6 тхпорпиридазинил-(4) изопро дают до 2590 и в течение 1 мн при пиптиокарбоната. Выщод 100.. сильном перемешивании добавляют 45...
Предыдущий патент: Устройство для обработки перемещаемого полотна материала
Следующий патент: Котел
Случайный патент: Устройство для дистанционного обнаружения объекта, скрытого под одеждой человека