Трициклические 5, 6-дигидро-9Н-пиразоло[3, 4-с]-1, 2, 4-триазоло[4, 3-a]пиридины, способ ингибирования фосфодиэстеразы типа IV, фармацевтическая композиция для лечения бронхиальной астмы.





Текст
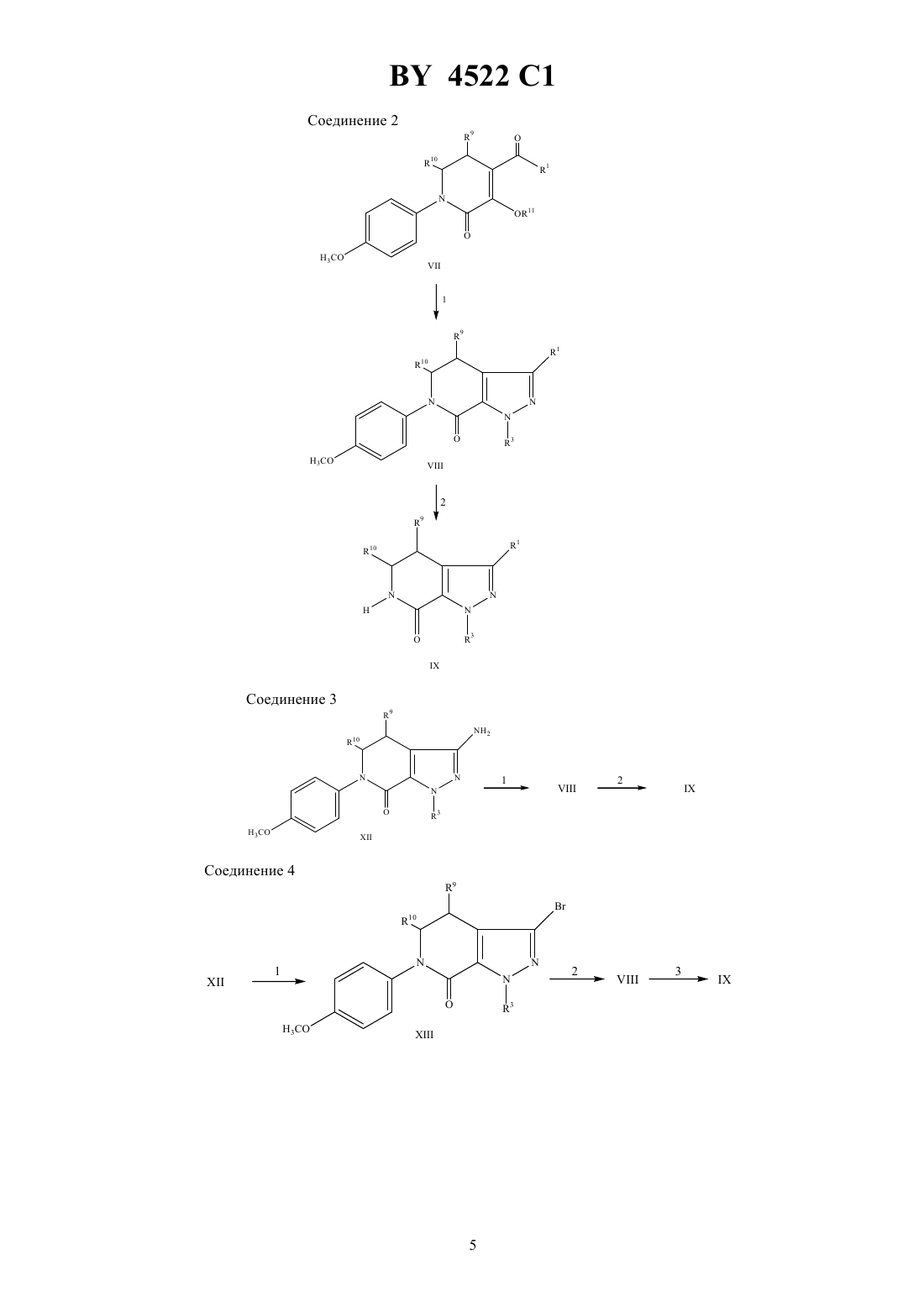
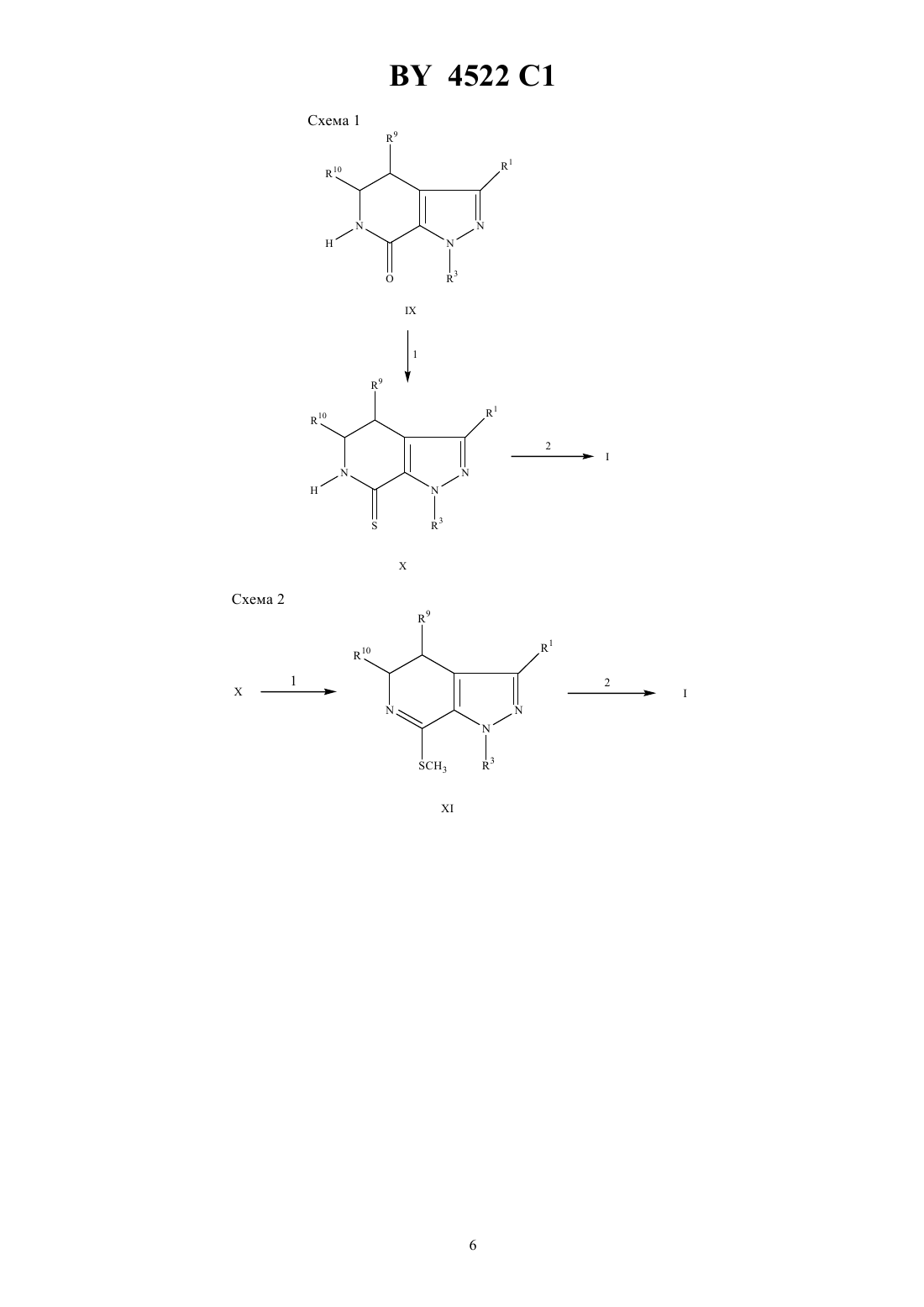
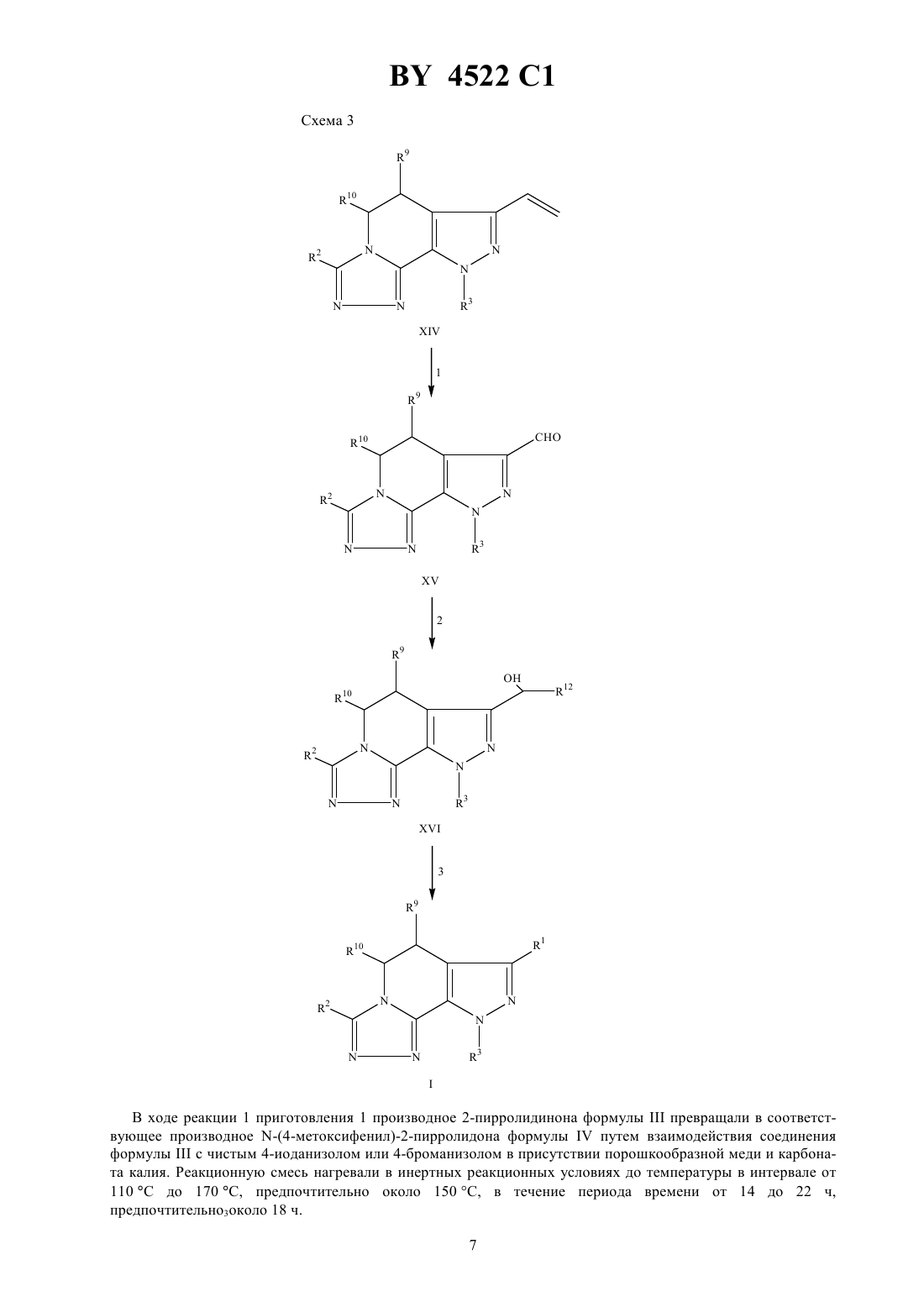
НАЦИОНАЛЬНЫЙ ЦЕНТР ИНТЕЛЛЕКТУАЛЬНОЙ СОБСТВЕННОСТИ ТРИЦИКЛИЧЕСКИЕ 5,6-ДИГИДРО-9 Н-ПИРАЗОЛО 3,4-с-1,2,4 ТРИАЗОЛО 4,3-ПИРИДИНЫ, СПОСОБ ИНГИБИРОВАНИЯ ФОСФОДИЭСТЕРАЗЫ ТИПА , ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ БРОНХИАЛЬНОЙ АСТМЫ где 1 - водород или С 1-С 6-алкил 2 - водород, С 1-С 6-алкил, С 3-С 7-циклоалкил, С 3-С 7-циклоалкил-С 1-С 2-алкил, насыщенная или ненасыщенная С 4-С 7-гетероцикло-(СН 2)-группа, где- 0, 1 или 2, содержащая в качестве гетероатома атом кислорода, серы или азота, причем, если в качестве гетероатома содержится атом азота, то кольцо дополнительно может включать группу 4, где 4 представляет собой водород или С 1-С 4-алкил или группа формулы где а - 1 или 2- 0 или 1 с - 0 или 1 5 - водород, гидрокси, С 1-С 6-алкил, С 1-С 3-алкокси, галоген или трифторметил- - - С 1-С 3-алкилен причем гетероциклическая группа может быть замещена С 1-С 2-алкилом, трифторметилом или галогеном 3 - С 3-С 7-циклоалкил-0-2-алкил или фенил, возможно замещенные С 1-С 2-алкилом, трифторметилом или галогеном 9 и 10 - независимо друг от друга - водород или С 1-С 6-алкил или их фармацевтически приемлемые соли. 2. Соединение по п. 1, где 1 - метил, этил или изопропил. 3. Соединение по п. 2, где 1 - этил. 4. Соединение по п. 1, где 3 - циклопентил. 5. Соединение по п. 1, где 1 - этил 3 - циклопентил 9 и 10 - водород. 6. Соединение по п. 1, выбранное из группы, включающей 9-циклопентил-5,6-дигидро-7-этил-3-фенил-9 Н-пиразоло 3,41,2,4-триазоло 4,3-пиридин 9-циклопентил-5,6-дигидро-7-этил-3-(4-пиридил)-9 Н-пиразоло 3,41,2,4-триазоло 4,3-пиридин 4522 1 9-циклопентил-5,6-дигидро-7-этил-3-(3-тенил)-9 Н-пиразоло 3,41,2,4-триазоло 4,3-пиридин 3,9-дициклопентил-5,6-дигидро-7-этил-9 Н-пиразоло 3,41,2,4-триазоло 4,3-пиридин 9-циклопентил-5,6-дигидро-7-этил-3-(1-метилциклогекс-1-ил)-9 Н-пиразоло 3,41,2,4-триазоло 4,3-пиридин 3-(трет-бутил)-9-циклопентил-5,6-дигидро-7-этил-9 Н-пиразоло 3,41,2,4-триазоло 4,3-пиридин 9-циклопентил-5,6-дигидро-7-этил-3-(тиен-2-ил)-9 Н-пиразоло 3,41,2,4-триазоло 4,3-пиридин 3-(2-хлорфенил)-9-циклопентил-5,6-дигидро-7-этил-9 Н-пиразоло 3,41,2,4-триазоло 4,3-пиридин 9-циклопентил-5,6-дигидро-7-этил-3-(2-йодфенил)-9 Н-пиразоло 3,41,2,4-триазоло 4,3-пиридин и 5,6-дигидро-7-этил-9-(4-фторфенил)-3-(1-метилциклогекс-1-ил)-9 Н-пиразоло 3,41,2,4-триазоло 4,3-пиридин. 7. Способ ингибирования фосфодиэстеразытипа , включающий введение пациенту эффективного количества соединения по п. 1. 8. Фармацевтическая композиция для лечения бронхиальной астмы, содержащая эффективное количество соединения по п. 1 и фармацевтически приемлемый носитель.(56)95/01980 1.1048746 , 1987.0429987 2, 1991.0343832 2, 1989. Настоящее изобретение относится к трициклическим 5,6-дигидро-9 Н-пиразоло 3,4-с-1,2,4-триазоло 4,3 апиридинам, которые обладают избирательным ингибирующим действием в отношении фосфодиэстеразы типаили фактора опухолеспецифического некрозаи поэтому эффективны при лечении астмы, артрита, бронхита, хронической обструкции дыхательных путей, псориаза, аллергического ринита, дерматита и других воспалительных заболеваний, таких как СПИД, сепсис, септический шок и другие заболевания, в частности кахексия, вызывающие образование фактора опухолеспецифического некроза. Соединения по настоящему изобретению способны ингибировать фосфодиэстеразу типаи фактор опухолеспецифического некроза. Настоящее изобретение относится также к фармацевтической композиции, предназначенной для лечения вышеуказанных заболеваний у млекопитающих, в частности у людей. После того, как стало известно, что циклическая адениловая кислотаявляется вторичным межклеточным мессенджером ( , . ., 1960, 12, 265), ученые стали уделять большое внимание ингибированию фосфодиэстеразы и терапевтическому воздействию на процессы, вызываемые этим веществом. Совсем недавно были обнаружены различные типы фосфодиэстеразы ( , , 1990, 11, 150), и избирательное ингибирование этих веществ сделало фармакотерапию более эффективной ( ,,. , 1991, 12, 19). В частности, было установлено, что ингибирование фосфодиэстеразы типаспособствует ингибированию медиатора воспаления( , др.,. Се ., 1989, 12 (приложение ),61) и расслаблению гладкой мышцы дыхательных путей (Т -/ Под ред. О, ,1988, 37, - ). Таким образом, соединения, которые ингибируют фосфодиэстеразу типа , но плохо воздействуют на другие типы фосфодиэстеразы, способны ингибировать медиаторы воспаления и расслаблять гладкую мышцу дыхательных путей, не воздействуя при этом на сердечно-сосудистую и не вызывая антитромбоцитарного действия. Известно, что фактор опухолеспецифического некроза является причиной возникновения многих инфекционных и аутоиммунных заболеваний, включая кахесию ( ., 1991, 285, 199). Кроме того, было установлено, что фактор опухолеспецифического некроза является первичным медиатором воспалительной реакции, наблюдаемой при сепсисе и септическом шоке (роо С.Е. и др., 1992, 62,11). Настоящее изобретение относится к соединению формулы 9 и его фармацевтически приемлемым солям,где 1 - водород, (С 1-6)алкил, (1-6)алкокси, (2-4)алкенил, фенил, диметиламино, (3-6)циклоалкил,(3-6)циклоалкил (1-3)алкил или (1-С 6)ацил, где алкильная, фенильная или алкенильная группы могут 2 4522 1 быть замещены одним или двумя гидрокси (С 1-3)алкильными или трифторметильными группами или одним-тремя галогенами 2 и 3, независимо друг от друга, выбраны из группы, включающей водород, (1-14)алкил, (17)алкокси(1-7)алкил, (2-14)алкенил, (3-7)циклоалкил, (3-7)циклоалкил(С 1-С 2)алкил, насыщенную или ненасыщенную (4-7) гетероциклическую(СН 2) группу, гдеравно 0,1 или 2, которая содержит в качестве гетероатома один или два элемента из группы, включающей кислород, серу, сульфонил, азот или 4,где 4 является водородом или (С 1-С 4) алкилом или группу формулы где а является целым числом от 1 до 5 и с равняются 0 или 1 5 представляет собой водород, гидрокси,(1-5)алкил, (С 2-С 5)алкенил, (С 1-С 5)алкокси, (3-6)циклоалкокси, галоген, трифторметил, 26,67, 67, 2 или 267, независимо друг от друга, являются водородом или (С 1-С 4)алкилом- кислород, сера, 2, СО или 8, где 8 является водородом или (1-С 4)алкилом, и -(С 1-5)алкилен или(С 2-С 6)алкенил, необязательно замещенный одной или двумя (1-7)алкильными или (С 3 С 7)циклоалкильными группами где каждая алкильная, алкенильная, циклоалкильная, алкоксиалкильная или гетероциклическая группы могут быть замещены одним-четырнадцатью, предпочтительно одним-пятью элементами, выбранными из группы, включающей (С 1-С 2)алкил, трифторметил или галоген 9 и 10, независимо друг от друга, выбраны из группы, включающей водород, (С 1-6)алкил, (16)алкокси, (С 6-С 10) арил и (С 6-С 10)арилокси. Термин алкил, если нет специального указания, означает насыщенные одновалентные углеводородные радикалы, отдельные части которых могут иметь прямые или разветвленные цепи или быть циклическими,либо представлять собой сочетание вышеуказанных типов. Термин алкокси означает -алкильные группы, в которых алкил имеет вышеуказанные значения. Термин тенил, если нет специального указания, означает тиофен-СН 2-. Термин арил, если нет специального указания, означает органический радикал, получаемый из ароматического углеводорода путем удаления одного атома водорода, в частности, фенил или нафтил, необязательно замещенный 1-3 заместителями, независимо друг от друга, выбранными из группы, включающей фтор, хлор, циано, нитро, трифторметил, (1-С 6)алкокси, (6-10)арилокси, трифторметокси, дифторметокси и (1-С 6)алкил. Термин арилокси означает -арильные группы, в которых арил имеет вышеуказанные значения. Термин ацил, если нет специального указания, означает радикал общей формулы , где- алкил,алкокси, арил, арилалкил или арилалкилокси, а термины алкил или арил имеют вышеуказанные значения. Предпочтительными соединения формулыявляются такие соединения, где 1 представляет собой метил, этил или изопропил. Другими предпочтительными соединениями формулыявляются такие соединения, где 3 представляет собой (1-6)алкил, (2-6)алкенил, (3-7)циклоалкил(1-С 6)алкил или фенил, необязательно замещенные 1 или 2 заместителями, выбранными из группы, включающей водород, гидрокси, (1-5)алкил, (2-5)алкенил,(1-5)алкокси, галоген, трифторметил, 26, 67, 67, 2 или 267, где 6 и 7, независимо друг от друга, представляют собой водород или (С 1-С 4)алкил. Наиболее предпочтительными соединениями формулыявляются следующие соединения 9-циклопентил-5,6-дигидро-7-этил-3-фенил-9 Н-пиразоло-3,4-с-1,2,4-триазоло 4,3-апиридин 9-циклопентил-5,6-дигидро-7-этил-3-(фуран-2-ил)-9 Н-пиразоло-3,4-с-1,2,4-триазоло 4,3-апиридин 9-циклопентил-5,6-дигидро-7-этил-3-(2-пиридил)-9 Н-пиразоло-3,4-с-1,2,4-триазоло 4,3-пиридин 9-циклопентил-5,6-дигидро-7-этил-3-(4-пиридил)-9 Н-пиразоло-3,4-с-1,2,4-триазоло 4,3-апиридин 9-циклопентил-5,6-дигидро-7-этил-3-(3-тенил)-9 Н-пиразоло 3,4-с-1,2,4-триазоло 4,3-апиридин 3-бензил-9-циклопентил-5,6-дигидро-7-этил-9 Н-пиразоло 3,4-с-1,2,4-триазоло 4,3-апиридин 9-циклопентил-5,6-дигидро-7-этил-3-пропил-9 Н-пиразоло 3,41,2,4-триазоло 4,3-апиридин 3,9-дициклопентил-5,6-дигидро-7-этил-9 Н-пиразоло 3,41,2,4-триазоло 4,3-пиридин 9-циклопентил-5,6-дигидро-7-этил-3-(1-метилциклогекс-1-ил)-9 Н-пиразоло 3,4-с-1,2,4-триазоло 4,3 пиридин 3-(трет-бутил)-9-циклопентил-5,6-дигидро-7-этил-9 Н-пиразоло 3,4-с-1,2, 4-триазоло 4,3-апиридин 9-циклопентил-5,6-дигидро-7-этил-3-(2-метилфенил)-9 Н-пиразоло 3,4-с-1,2,4-триазоло 4,3-пиридин 9-циклопентил-5,6-дигидро-7-этил-3-(2-метоксифенил)-9-пиразоло 3,4-с-1,2,4-триазоло 4,3-апиридин 9-циклопентил-5,6-дигидро-7-этил-3-(тиен-2-ил)-9 Н-пиразоло 3,4-с-1,2, 4-триазоло 4,3-пиридин 3-(2-хлорфенил)-9-циклопентил-5,6-дигидро-7-этил-9 Н-пиразоло 3,41,2,4-триазоло 4,3-пиридин 3 4522 1 9-циклопентил-5,6-дигидро-7-этил-3-(2-иодфенил)-9 Н-пиразоло 3,41,2,4-триазоло 4,3-апиридин 9-циклопентил-5,6-дигидро-7-этил-3-(2-трифторметилфенил)-9-пиразоло 3,4-с-1,2,4-триазоло 4,3 пиридин 5,6-дигидро-7-этил-9-(4-фторфенил)-3-(1-метилциклогекс-1-ил)-9 Н-пиразоло 3,4-с-1,2,4-триазоло 4, 3 апиридин. Настоящее изобретение относится также к способу ингибирования фосфодиэстеразы типаи фактора опухолеспецифического некроза, в соответствии с которым больному вводят эффективное количество соединения формулыили его фармацевтически приемлемой соли. Настоящее изобретение относится также к фармацевтической композиции, предназначенной для (а) лечения астмы, артрита, бронхита, хронической обструкции дыхательных путей, псориаза, аллергического ринита, дерматита и других воспалительных заболеваний, вызываемых фосфодиэстеразой типа , СПИДа, сепсиса, септического шока и других заболеваний, таких как кахексия, вызываемых фактором опухолеспецифического некроза илиингибирования фосфодиэстразы типаи фактора опухолеспецифического некроза, который содержит эффективное количество соединения формулыили его фармацевтически приемлемые соли вместе с фармацевтически приемлемым носителем. Данное изобретение относится к способу профилактики или лечения одного из таких заболеваний, как астма, артрит, бронхит, хроническая обструкция дыхательных путей, псориаз, аллергический ринит, дерматит и другие воспалительные заболевания, СПИД, септический шок и другие заболевания, в частности кахексия, вызывающие образование фактора опухолеспецифического некроза, в соответствии с которым больному вводят эффективное количество соединения формулыили его фармацевтически приемлемой соли. Приведенные ниже схемы реакций иллюстрируют, но не ограничивают способы получения соединений по настоящему изобретению. Если нет специального указания, 1, 2, 3, 9 и 10 в этих схемах реакций и в описании изобретения имеют вышеуказанные значения. Соединение 1 В ходе реакции 1 приготовления 1 производное 2-пирролидинона формулыпревращали в соответствующее производное -(4-метоксифенил)-2-пирролидона формулыпутем взаимодействия соединения формулыс чистым 4-иоданизолом или 4-броманизолом в присутствии порошкообразной меди и карбоната калия. Реакционную смесь нагревали в инертных реакционных условиях до температуры в интервале от 110 С до 170 С, предпочтительно около 150 С, в течение периода времени от 14 до 22 ч,предпочтительно 3 около 18 ч. 7 4522 1 В ходе реакции 2 приготовления 1 к суспензии магния в безводном апротонном растворителе добавляли 1 галогенид, где 1 является (1-С 6)алкилом. Реакционную смесь нагревали с обратным холодильником до полного расхода магния, после чего суспензию охлаждали до температуры от около -15 С до около 15 С,предпочтительно до 0 С. Затем добавляли производное -(4-метоксифенил)-2-пирролидона формулыи нагревали реакционную смесь до комнатной температуры при одновременном перемещении в течение периода времени от около 1,5 до около 2,5 ч, предпочтительно в течение около 2 ч. Предпочтительными алкилгалогенидами являются бромметан, бромэтан или бромпропан. Предпочтительным безводным апротонным растворителем является безводный простой эфир. Желаемый промежуточный продукт выделяли и превращали его в соответствующее производное 1,2,5,6 тетрагидропиридина формулыпутем диспергирования осадка в смеси неполярного апротонного растворителя и основания. Добавляли этилоксалилхлорид и нагревали реакционную смесь с обратным холодильником в течение периода времени от около 1,5 ч до около 4,5 ч, предпочтительно в течение 3,0 ч. Предпочтительным неполярным апротонным растворителем является бензол, а предпочтительным основанием гидроксид натрия. Растворители удаляли, а полученный остаток обрабатывали раствором алкоксида натрия в этаноле. После нагревания с обратным холодильником в течение периода времени от 1 до 3 ч, предпочтительно в течение 1,5 ч, смесь концентрировали при пониженном давлении и подкисляли до рН с помощью хлористоводородной кислоты. В ходе реакции 3 приготовления 1 производное формулыпревращали в соответствующее производное 3-метокси-1,2,5,6-тетрагидропиридина формулыпутем нагревания с обратным холодильником реакционной смеси производного формулыи 3-метил-1-р-толилтриазена в апротонном растворителе, предпочтительно 1,2-дихлорэтане, в течение периода времени от около 30 мин до около 2 ч, предпочтительно в течение около 45 мин. В ходе реакции 1 приготовления 2 производное 1,2,5,6-тетрагидропиридина формулы , где 11 является водородом или метилом, превращали в соответствующее производное 4,5,6,7-тетрагидро-7-оксо-1 Нпиразоло 3,4-спиридина формулыпутем взаимодействия с соединением гидразина формулы 3 НН 2,где 3 имеет вышеуказанные значения. Оба производные соединения формулы , 3-гидрокси и 3-метокси,можно использовать в качестве исходных материалов при осуществлении реакции одного из трех типов. В одних представленных реакционных условиях производное 1,2,5,6-тетрагидропиридина формулыпревращали в соответствующее соединение формулыпутем взаимодействия соединенияс гидрохлоридом гидразина и алкоксидом натрия в безводном полярном протонном растворителе. Предпочтительным алкоксидом натрия является метоксид натрия, а предпочтительным безводным полярным протонным растворителем - безводный этанол. Реакционную смесь нагревали с обратным холодильником в течение периода времени от около 9 до около 15 ч, предпочтительно в течение около 12 ч. При других указанных реакционных условиях производное 1,2,5,6-тетрагидропиридина формулыпревращали в соответствующее соединение формулыпутем взаимодействия соединенияс гидразином в безводном полярном протонном растворителе, предпочтительно этаноле. Реакционную смесь нагревали с обратным холодильником в течение периода времени от около 16 до около 24 ч, предпочтительно в течение 20 ч. В третьих указанных реакционных условиях производное 1,2,5,-тетрагидропиридина формулыпревращали в соответствующее соединение формулыпутем взаимодействия соединенияс гидразином или гидрохлоридом гидразина в полярном протонном растворителе, предпочтительно метаноле. Реакционную смесь нагревали до температуры от около 70 С до около 110 С, предпочтительно до около 90 С, в слабом потоке азота до полного удаления растворителя. Чистую смесь затем нагревали до температуры от около 120 С до около 180 С, предпочтительно до около 150 С, в течение периода времени от около 30 до около 90 мин, предпочтительно в течение около 60 мин. В ходе реакции 2 приготовления 2 производное формулыпревращали в соответствующее соединение 6-Н-4,5,6,7-тетрагидро-7-оксо-1 Н-пиразоло 3,4-спиридина формулыпутем взаимодействия раствора производного формулыв полярном апротонном растворителе, предпочтительно, ацетонитриле, с раствором нитрата церияи аммония в воде при температуре от около -15 С до 15 С, предпочтительно при около 0 С, в течение периода времени от около 20 до около 50 мин, предпочтительно в течение около 35 мин. После окончания реакции эту смесь разбавляли водой и экстрагировали этилацетатом. Объединенные органические продукты промывали сначала насыщенным раствором бикарбоната натрия, а потом сульфитом натрия. В ходе реакции 1 приготовления 3 производное формулы , полученное в соответствии с описанием,приведенным в патенте США 3423414, превращали в соответствующее производное формулы , где 1 является диметиламино, путем обработки соединениягидридом натрия в полярном апротонном растворителе, таком как тетрагидрофуран, при температуре от около 0 С до около 62 С, предпочтительно при около 25 С в течение периода времени от около 1 до около 6 ч, предпочтительно в течение около 1 ч. При комнатной температуре в реакционную смесь добавляли избыточное количество метилиодида, после чего 4522 1 эту смесь перемешивали в течение периода времени от около 1 до около 24 ч, предпочтительно в течение около 2 ч. В ходе реакции 2 приготовления 3 производное формулыподвергали дальнейшему взаимодействию с образованием соответствующего соединения 6-Н-4,5,6,7-тетрагидро-7-1 Н-пиразоло 3,4-пиридина формулы , в которой 1 является диалкиламино, в соответствии со способом, описанным выше для реакции 2 приготовления 2. В ходе реакции 1 приготовления 4 производное формулыпревращали в соответствующее производное формулыпутем взаимодействия соединенияс бромтриметилсиланом и нитритом натрия в апротонном растворителе, таком как тетрахлорметан, при температуре от около 0 С до около 25 С, предпочтительно при около 25 С, в течение периода времени от около 6 до около 48 ч, предпочтительно в течение около 24 ч. В ходе реакции 2 приготовления 4 соединение формулыпревращали в соответствующее соединение формулы , где 1 является винилом, путем взаимодействия соединенияс винилтрибутиловым и каталитическим количеством тетракис(трифенилфосфин)палладия(О) в неполярном апротонном растворителе,таком как бензол, при температуре от около 80 С до около 120 С, предпочтительно при около 100 С, в течение периода времени от около 24 до около 72 ч, предпочтительно в течение около 48 ч. В ходе реакции 3 приготовления 3 производное формулыподвергали дальнейшему взаимодействию с образованием соответствующего соединения 6-Н-4,5,6,7-тетрагидро-7-оксо-1 Н-пиразоло 3,4-пиридина формулы , в которой 1 является алкенилом, в соответствии со способом, описанным выше для реакции 2 приготовления 2. В ходе реакции 1 приготовления 1 производное лактама формулыпревращали в соответствующее производное тиолактама формулыпутем взаимодействия производногос пентасульфидом фосфора в полярном апротонном растворителе, таком как 1,4-диоксан или пиридин. Реакционную смесь нагревали с обратным холодильником в течение периода от около 12 до около 48 ч, предпочтительно в течение около 18 ч. В ходе реакции 2 приготовления 1 тиолактам формулыпревращали в соответствующее производное трициклического 5,6-дигидро-9 Н-пиразоло 3,4-с-1,2,4-триазоло 4,3-апиридина формулыпутем обработки соединениябезводным гидразином в присутствии безводного апротонного растворителя, такого как пиридин, в нейтральных условиях реакции. Реакционную смесь нагревали до температуры от около 50 С до около 100 С, предпочтительно до около 70 С, в течение периода времени от около 5 до около 30 мин, предпочтительно в течение около 5 мин. Летучие вещества удаляли при пониженном давлении, после чего добавляли свежийбезводный апротонный растворитель, предпочтительно пиридин, а затем апротонный хлорангидрид формулы 2, где 2 имеет вышеуказанные значения. Полученную реакционную смесь перемешивали в течение периода времени от около 1 до около 4 ч, предпочтительно в течение около 2 ч. Летучие вещества еще раз удаляли при пониженном давлении. Остаток растворяли в апротонном растворителе,таком как диметилформамид, и нагревали с обратным холодильником в течение периода времени от около 1 до около 4 ч, предпочтительно в течение около 2 ч. В ходе реакции 1 приготовления 2 производное тиолактама формулыпревращали в соответствующее метилтиосоединение формулыпутем обработки смеси соединенияи силикагеля в апротонном растворителе, таком как простой эфир, раствором диазометана в простом эфире. Температура реакции должна находиться в интервале от около -5 С до около 10 С, предпочтительно реакция осуществляется при температуре около 0 С в течение периода времени от около 30 мин до около 5 ч, предпочтительно в течение около 1 ч. В ходе реакции 2 приготовления 2 производное метилтиола формулыпревращали в соответствующее соединение трициклического 5,6-дигидро-9 Н-пиразоло 3,41,2,4-триазоло 4,3-пиридина формулыпутем взаимодействия соединенияс производным гидразида формулы 22 или с соответствующей хлористоводородной солью в апротонном растворителе, таком как пиридин, в нейтральных условиях реакции. Реакционную смесь нагревали до температуры от около 120 С до около 150 С, предпочтительно до около 135 С, в течение периода времени от около 2 до около 6 ч, предпочтительно в течение около 4 ч. Летучие вещества удаляли при пониженном давлении, а полученное масло продолжали нагревать до температуры от около 135 С до около 165 С, предпочтительно до около 150 С, в течение периода времени от около 2 до около 6 ч,предпочтительно в течение около 4 ч. В ходе реакции 1 приготовления 3 производное формулы Х превращали в соответствующее производное формулы Х в соответствии со способом, описанным в журнале, 33,1714 (1955). В ходе реакции 2 приготовления 3 производного формулы Х превращали в соответствующее производное формулы Х, где 12 является (1-С 6)алкилом, путем взаимодействия соединения Х с алкиллитием в полярном апротонном растворителе, таком как простой эфир, при температуре от около -50 С до около -80 С, предпочтительно при около -78 С, в течение периода времени от около 15 мин до около 2 ч, предпочтительно в течение около 30 мин. 9 4522 1 В ходе реакции 3 приготовления 3 производное формулы Х превращали в соответствующее производное 5,6-дигидро-9 Н-пиразоло 3,4-с-1,2,4-триазоло 4,3-апиридина формулы , где 1 является (С 1-6) ацилом, путем обработки соединения Х хлорохроматом пиридиния в неполярном апротонном растворителе, таком как метиленхлорид, при комнатной температуре в течение периода времени от около 6 до около 24 ч, предпочтительно в течение около 12 ч. Результаты следующего анализаподтверждают способность этих соединений или их фармацевтически приемлемых солей ингибировать фосфодиэстеразу(4) и эффективно лечить воспалительные заболевания. Биологические анализы. Фосфодиэстераза , вызывающая эозинофильный лейкоцитоз у человека. Кровь из периферической кровеносной системы человека помещали в этилендиаминтетрауксусную кислоту,разбавляли 12, используя для этого буфер, представляющий собой пиперазин-,-бис-2-этан-сульфокислоту, а затем расслаивали с помощью раствора перколла. Градиенты формировали путем центрифугирования,осуществляемого в течение 30 мин со скоростью 2000 оборотов в минуту при температуре 4 С. Остальную часть процедуры выделения на основе метода, предложенного а и др., . , 152, 5457 (1994), выполняли при температуре 4 С. Слой нейрофилов и эозинофилов собирали из градиента персолла, а эритроциты лизировали. Остальные клетки промывали пиперазин-,-бис-этансульфокислотой (1 околоплодной сыворотки теленка),инкубировали вместе с микрошариками анти-С 16 в течение 1 ч и пропускали через колонку из ферромагнетика с целью удаления нейрофилов. Эозинофилы собирали в элюате и определяли их жизнеспособность с помощью трипана синего и чистоту с помощью красителя, обеспечивающего быструю дифференциацию. При использовании этого метода чистота эозинофилов превышала 99 . Очищенные эозинофилы повторно суспендировали в 750 мкл буфера для лизиса фосфодиэстеразы (20 ммолей триэтиламина, 1 ммоль этилендиаминтетрауксусной кислоты, 100 мкг/мл бацитрацина, 2 ммоля бензамида, 50 мкмолей лейпептина, 50 мкмолей фенилметилсульфонилфторида, 100 мкг/мл ингибитора соевого трипсина) и быстро замораживали в жидком азоте. Клетки медленно оттаивали и разрушали ультразвуком. Мембраны интенсивно перемешивали (измельчение клеток подтверждали окрашиванием фрагментов трипаном синим). Измельченные клетки центрифугировали для удаления мембран со скоростью 45000 об./мин в течение 30 мин при температуре 4 С. Цитозоль декантировали, а мембрану повторно суспендировали до 200 мкг/мл для использования в качестве источника фосфодиэстразы при выполнении гидролизного анализа, в результате которого было получено дискриминационное окно, обеспечивающее от 3000 до 5000 подсчетов. Соединения растворяли в диметилсульфоксиде в количестве 10-2 моля, а затем разбавляли водой в отношении 125 до достижения 410-4 раствора. Эту суспензию последовательно разбавляли 4 диметилсульфоксидом в отношении 110 так, что окончательная концентрация диметилсульфоксида в пробе равнялась 1 . Анализ ингибирования фосфодиэстеразы. В стеклянные пробирки размером 1275 мм помещали 25 мкл буфера для анализа фосфодиэстеразы (200 ммолей трис/40 ммолей 12) 25 мкм 4 нмоля/мл аденозин-3,5-циклофосфата 25 мкл испытуемого соединения 25 мкл источника фосфодиэстеразы (мембрана). Фоновая контрольная пробамембрана после кипячения в течение 10 мин. Положительная контрольная проба 25 мкл некипяченой мембраны. Инкубация в течение 25 мин в водяной бане при температуре 37 С. Реакцию прекращали после кипячения проб в течение 5 мин. Пробы вводили в колонку из аффи-геля (1 мл слой),предварительно уравновешенную 0,25 раствором уксусной кислоты, после чего добавляли 0,1 ммоля -2 гидроксиэтилпиперазин 2-этансульфокислоты и 0,1 ммоля промывочного буфера на основе (рН 8,5). Аденозин-3,5 циклофосфат вымывали из колонки с помощью смеси -(2-гидроксиэтил)пиперазин 2-этансульфокислоты и , аденозин-5-монофосфат элюировали 0,25 раствором уксусной кислоты в виде 4 мл объемов. Для подсчета использовали 1 мл элюата в 3 мл сцинтилляционной жидкости, выполняя эту процедуру в течение 1 мин 3. Превращение субстрата(число подсчетов в минуту у положительной контрольной пробы 4)/общую активность. Степень превращения должна составлять от 3 до 15 , чтобы результаты эксперимента можно было считать действительными.ингибирования 1-(число подсчетов в минуту у элюированной пробы - число подсчетов в минуту у фоновой пробы/число подсчетов в минуту у контрольной пробы - число подсчетов в минуту у фоновой пробы)100. 50 выводили путем линейной регрессии титрационной кривой ингибирования (линейная часть) и выражали в мкмолях. Фактор опухолеспецифического некроза. Результаты следующего анализаподтверждают способность соединений по настоящему изобретению или их фармацевтически приемлемых солей ингибировать образование фактора опухолеспецифиче 10 4522 1 ского некроза и эффективно лечить заболевания, вызывающие образование фактора опухолеспецифического некроза. Кровь из периферической кровеносной системы (100 мл), взятую у добровольцев, помещали в этилендиаминтетрауксусную кислоту. Мононуклеарные клетки отделяли с помощью смеси фиколла и гипака и трижды промывали не полностью сбалансированным солевым раствором Ханкса. Клетки повторно суспендировали с достижением конечной концентрации, равной 1106 клеткам на мл в предварительно нагретом растворе, содержащем 5 околоплодной сыворотки теленка, глутамин и нистатин). Моноциты помещали в количестве 1106 клеток в 1,0 мл на планшеты с 24 ячейками. Клетки инкубировали при температуре 37 С (5 диоксида углерода) и оставляли на 2 ч для прилипания к планшетам, после чего неприлипшие клетки удаляли осторожным промыванием. Испытуемые соединения (10 мкл) добавляли к клеткам в количестве, которое в 3-4 раза превышало концентрацию клеток, и инкубировали в течение 1 ч. В соответствующие ячейки добавляли липополисахарид (10 мкл). Планшеты инкубировали в течение ночи (18 ч) при температуре 37 С. В конце инкубационного периода фактор опухолеспецифического некроза анализировали с помощью твердофазного иммуноферментного анализа (комплект.). Определения 50 производили отдельно для каждого соединения на основе анализа линейной регрессии. Фармацевтически приемлемые соли присоединения кислоты соединений по настоящему изобретению включают соли, образуемые с такими кислотами, как , , 3, 24, 34, 33, 3643, 32, глюконовая кислота, винная кислота, малеиновая кислота и янтарная кислота, но не ограничиваются ими. Фармацевтически приемлемые катионные соли соединений формулыпо настоящему изобретению, в которой 5 является 26 и 6 - водородом, включают соли натрия, калия, кальция,магния, аммония, ,-дибензилэтилендиамина, -метилглюкамина (меглюмина), этаноламина и диэтаноламина, но не ограничиваются ими. Дозировка соединений формулыи их фармацевтически приемлемых солей (далее именуемых активными соединениями по настоящему изобретению), предназначенных для перорального введения в профилактических целях или для лечения воспалительных заболеваний, составляет 0,1-400 мг в сутки для взрослого больного средней комплекции(70 кг). Таким образом, предназначенные для типичного взрослого больного, таблетки или капсулы содержат от 0,1 до 50 мг активного соединения в фармацевтически приемлемом наполнителе или носителе. Дозировка лекарственных средств для внутривенного вливания обычно составляет от 0,1 до 40 мг активного соединения на одну дозу. Дозировка лекарственных средств для введения через нос или для ингаляции обычно производится с использованием раствора,содержащего от 0,1 до 1 активного вещества (процентное соотношение объемов). Врач на практике определяет необходимую дозировку, которая более всего подходит определенному больному, поэтому она может изменяться в зависимости от возраста, веса и реакции больного. Вышеуказанные дозировки являются типичными, но, несомненно,могут иметь место случаи, требующие более высокой или более низкой дозировки, поэтому необходимо отметить, что все эти дозировки входят в объем настоящего изобретения. Составы по настоящему изобретению, предназначенные для ингибирования фактора опухолеспецифического некроза, можно вводить больным различными способами, включая пероральный, парентеральный и местный. Как правило, активное соединение вводят перорально или парентерально в дозах, составляющих от 0,1 до 25 мг/кг веса тела больного в сутки, предпочтительно от 0,3 до 5 мг/кг. Соединение формулыможно применять местно в виде мази или крема с концентрацией от 0,5 до 1 путем нанесения на больное место 2 или 3 раза в день. Однако дозировка будет несколько отличаться в зависимости от состояния больного. Специалист в любом случае должен самостоятельно определить нужную дозу для каждого больного. Активные соединения по настоящему изобретению можно вводить больным отдельно, но обычно они используются в смеси с фармацевтическим разбавителем или носителем, выбираемым в зависимости от предполагаемого способа введения и обычной фармацевтической практики. Например, для перорального введения можно использовать таблетки, содержащие такие наполнители, как крахмал или лактоза капсулы или лепешки, содержащие только активные соединения или в смеси с наполнителями эликсиры или суспензии, содержащие ароматизаторы или красители. Активные соединения по настоящему изобретению можно вводить парентерально, например внутривенно, внутримышечно или подкожно. Для парентерального введения их лучше всего использовать в виде стерильного водного раствора, который может содержать и другие вещества например, достаточное количество солей или глюкозы, позволяющих сделать этот раствор изотоническим. Настоящее изобретение иллюстрируется следующими примерами, но не ограничивается ими. Исходные материалы, используемые в приготовлениях 1-4, получали в соответствии с описанием, приведенным в публикации 95/01980 по Договору о патентной кооперации. Приготовление 1. 1-Циклопентил-4,5-дигидро-3-этил-7-метилтио-1 Н-пиразоло 3,4-пиридин. Смесь 1-циклопентил-3-этил-7-тио-4,5,6,7-тетрагидро-1 Н-пиразоло 3,4-спиридина (0,322 г), нейтрального силикагеля (10 г) и простого эфира (100 мл) помещали в 500 колбу Эрленмейера, перемешивали с помощью магнитной мешалки и охлаждали до 0 С. В эту смесь медленно вводили избыточное количество раствора диазометана в простом эфире. Происходило выделение газа, и через 1 ч реакционную смесь резко охлаждали путем добавления уксусной кислоты (1 капля), фильтровали и концентрировали при пониженном давлении с образованием желтого масла. Это мас 11 4522 1 ло очищали посредством хроматографии на колонке из силикагеля с использованием в качестве элюента смеси этилацетата и гексана с соотношением 14, что позволило получить 0,232 г желтого масла. Результаты анализа, вычисленные для 14213 С, 63,85 , 8,04 , 15,94. Найдено С, 64,01 , 8,37 , 15,71. Приготовление 2. 1-циклопентил-3-этил-7-тио-4,5,6,7-тетрагидро-1 Н-пиразоло 3,4-спиридин. Раствор 1-циклопентил-3-этил-7-тио-4,5,6,7-тетрагидро-1 Н-пиразоло 3,4-спиридина (10,0 г) в безводном 1,4 диоксане обрабатывали пентасульфидом фосфора (3,9 г). После перемешивания при нагревании с обратным холодильником в течение 12 ч смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученное желтое масло растворяли в метиленхлориде и промывали водой и рассором, сушили над сульфатом натрия и концентрировали при пониженном давлении. Оранжевый остаток очищали посредством хроматографии на колонке из силикагеля с использованием в качестве элюента градиентной смеси гексанов в метиленхлориде, в результате чего было получено 9,3 г желтого твердого вещества. Температура плавления 152-153 С. Результаты анализа,вычисленные для 13193 С, 62,63 , 7,68 , 16,86. Найдено С, 62,14 , 7,51 , 16,35. Приготовление 3. 1-Циклопентил-3-этил-6-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1 Н-пиразоло 3,4-пиридин. Перемешиваемую смесь 3-метокси-1-(3-метоксифенил)-2-оксо-4-пропионил-1,2,5,6-тетрагидропиридина (0,49 г,16. ммоля), гидрохлорида циклопентилгидразина (0,40 г) и метоксида натрия (46 мг, 0,85 ммоля) в безводном этаноле нагревали с обратным холодильником. Через 16 ч смесь концентрировали при пониженном давлении и хроматографировали на колонке из силикагеля с использованием в качестве элюента смеси этилацетата и гексана с соотношением 14, что позволило получить белое твердое вещество. В результате перекристаллизации из простого эфира были получены белые иглы. Температура плавления 64-65 С масс-спектрометрия/340,2025 масс-спектрометрия с высоким разрешением 340,2046. Приготовление 4. 1-Циклопентил-3-этил-7-оксо-4,5,6,7-тетрагидро-1 Н-пиразоло 3,4-пиридин. Перемешиваемый раствор 1-циклопентил-3-этил-6-(4-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1 Нпиразоло 3,4-пиридина (2,58 г, 7,60 ммоля) в ацетонитриле (90 мл) с температурой 0 С обрабатывали раствором нитрата церия и аммония (12,5 г, 22,8 ммоля) в воде (110 мл). После перемешивания в течение 35 мин эту смесь разбавляли водой (550 мл) и экстрагировали этилацетатом (100 мл 4). Объединенные органические продукты промывали сначала 50 -ным насыщенным раствором бикарбоната натрия (250 мл), а потом 10 -ным раствором сульфита натрия до тех пор, пока промывочная вода не становилась бледно-желтой. Органический слой продолжали промывать насыщенным раствором бикарбоната и рассола и обрабатывали обесцвечивающим активированным углем. После перемешивания в течение 30 мин смесь сушили над сульфатом натрия, фильтровали через целит и концентрировали при пониженном давлении. Коричневый остаток перекристаллизовывали из простого эфира с образованием 0,814 г твердого дубильного вещества. Температура плавления 143-145 С масс-спектрометрия (/) 234 1 ЯМР(250 МГц, СС 3) 1,21 (т,7,6 Гц, 3), 1,62-2,13 (м, 8), 2,62 (к,7,6 Гц, 2), 2,73 (т,6,8 Гц, 2), 3,51 (двойной триплет,2,7 и 6,8 Гц, 2), 5,47 (с, 1), 5,61 (пентет,7,7 Гц, 1). Пример 1. 9-Циклопентил-5,6-дигидро-7-этил-3-(3-пиридил)-9 Н-пиразоло 3,41,2,4-триазоло 4,3-апиридин. 1-Циклопентил-4,5-дигидро-3-этил-7-метилтио-1 Н-пиразоло-3,4-пиридин (0,036 г, 0,14 ммоля) и гидразид никотиновой кислоты (0,021 г, 0,15 ммоля) растворяли в безводном пиридине (5 мл) в колбе, высушенной на пламени. Колбу подсоединяли к высушенному в печи конденсатору, который был закупорен уплотнительной мембраной и имел выход в барботер. Мембрану прокалывали длинной иглой из нержавеющей стали и вводили эту иглу в раствор,перемешиваемый с помощью магнитной мешалки. Через длинную иглу выходили пузырьки азота. Колбу нагревали до температуры 135 С в течение 4 ч. Пиридин удаляли в условиях продувки азотом. Полученное масло нагревали до 150 С в течение 4 ч. Колбу охлаждали до комнатной температуры и получали из нее 0,045 г указанного в заголовке неочищенного соединения в виде белого твердого вещества. Неочищенный продукт не содержал загрязняющих примесей, определяемых с помощью тонкослойной хроматографии. Этот продукт можно очистить посредством хроматографии на колонке из силикагеля с использованием в качестве элюента градиентной смеси этилацетата и гексана или путем перекристаллизации из смеси этилацетата в гексане. Температура плавления 140-145 С (неочищенный продукт) 1 ЯМР (300 МГц, 3)1,24 (т,7,6 Гц, 3), 1,72 (м, 2), 1,94 (м, 2), 2,16 (м, 4), 2,66 (к,7,6 Гц,2), 2,98 (т,7,0 Гц, 2), 4,25 (т,7,0 Гц, 2), 5,60 (квинтет,7,7 Гц, 1), 7,48 (двойной дублет,- 4,9 и 7,8 Гц,1), 8,05 (д,8,0 Гц, 1), 8,75 (двойной дублет,1,4 и 4,9 Гц, 1), 8,9 (д,1,7 Гц, 1). Результаты анализа, вычисленные для С 19 Н 226 С, 68,23 , 6,63 , 25,13. Найдено С, 67,39 , 6,87 , 24,00. Примеры 2-18. Осуществляя взаимодействие соответствующего гидразида с 1-циклопентил-4,5-дигидро-3-этил-7-метилтио-1 Нпиразоло 3,4-пиридином, в соответствии со способом, описанным в примере 1, были получены следующие соединения формулы , где 1 является этилом, а 3 - циклопентилом. Пример 19. 9-Циклопентил-5,6-дигидро-7-этил-3-(тиен-2-ил)-9 Н-пиразоло 3,41,2,4-триазоло 4, 3-пиридин. 1-Циклофенил-3-этил-7-тио-4,5,6,7-тетрагидро-1 Н-пиразоло 3,4 пиридин (0,35 г, 1,4 ммоля) растворяли в 4 мл безводного пиридина, находящегося под слоем азота в колбе, высушенной на пламени. Эту колбу нагревали до 70 С и добавляли 1,5 мл безводного гидразина. Желтый раствор становился розовым, после чего его перемешивали в течение 5 мин. Пиридин и избыток гидразина удаляли при пониженном давлении с образованием розового твердого вещества, которое становилось светло-зеленым после нахождения в вакууме (приблизительно 0,1 мм) в течение 30 мин. В эту колбу сначала добавляли безводный пиридин (4 мл), а затем 2 тиофенкарбонилхлорид (0,69 г, 4,7 ммоля) и перемешивали смесь в течение 2 ч. Пиридин удаляли при пониженном давлении, а остаток растворяли в диметилформамиде (4 мл) и нагревали с обратным холодильником в течение 2 ч. После этого смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали этилацетатом. Водный слой подщелачивали до рН 12 с помощью 1 н. раствора гидроксида натрия и трижды экстрагировали этилацетатом. Соединенные органические вещества промывали 1 н. раствором гидроксида натрия, водой и рассолом, сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученное масло очищали посредством хроматографии на колонке из силикагеля с использованием в качестве элюента градиентной смеси этилацетата и гексана, что позволило получить 304 мг указанного в заголовке соединения в виде белого твердого вещества. Температура плавления 125-126 С 1 ЯМР (300 МГц,3)1,25 (т,7,5 Гц, 3), 1,60-1,74 (м, 2), 1,9-2,0 (м, 2), 2,11-2,21 (м, 4), 2,67 (к,7,6 Гц, 2),3,00 (т,7,1 Гц, 2), 4,30 (т,7,1 Гц, 2), 5,60 (квинтет,7,7 Гц, 1), 7,20 (двойной дублет,3,9 и 5,1 Гц, 1), 7,49-7,54 (м, 2). Результаты анализа вычисл. для 18215 С, 63,68, , 6,24, , 20,63. Найдено С, 63,66, , 6,19, , 21,00. Примеры 20-30. В результате взаимодействия соответствующего хлорангидрида с гидразином и 1-циклопентил-3-этил-7 тио-4,5,6,7-тетрагидро-1 Н-пиразоло 3,4-пиридином, осуществляемого в соответствии с процедурой, описанной в примере 19, были получены следующие соединения формулы , в которой 1 является этилом,3 88-92 174-175 132-134 158 масло масло масло 161-163 145-147 154-155 144-145 Пример 31. 5,6-Дигидро-7-этил-9-(4-фторфенил)-3-(1-метилциклогекс-1-ил)-9 Н-пиразоло 3,4-с -1,2,4-триазоло 4,3 пиридин. 3-Этил-1(4-фторфенил-7-тио-4,5,6,7-тетрагидро-1 Н-пиразоло 3,4-спиридин (0,092 г) растворяли в безводном пиридине (5 мл) и нагревали полученный раствор до 70 С. Добавляли безводный гидразин (2 мл), после чего желтый раствор медленно становился бежевым. Через 5 мин при пониженном давлении удаляли летучие вещества, что позволило получить твердое вещество желтого цвета. Затем добавляли безводный пиридин (5 мл), а вслед за ним хлористый 1-метилцикло-гексанкарбонил (0,2 г). После перемешивания в течение 2 ч при комнатной температуре и пониженном давлении удаляли пиридин, а остаток растворяли в диметилформамиде (5 мл). После перемешивания при нагревании с обратным холодильником в течение 12 ч этот раствор охлаждали до комнатной температуры,разбавляли водой и экстагировали этилацетатом. Соединенные органические вещества промывали водой и рассолом, а затем сушили над сульфатом натрия. В результате концентрирования при пониженном давлении было получено светло-коричневое масло. Это масло очищали посредством хроматографии на колонке из силикагеля с использованием в качестве элюента смеси этилацетата и гексана с соотношением 12, в результате чего было получено 0,09 г светло-желтого твердого вещества. Температура плавления 60-61 С масс-спектрометрия (/) 380. Национальный центр интеллектуальной собственности. 220072, г. Минск, проспект Ф. Скорины, 66. 14
МПК / Метки
МПК: C07D 471/14, A61K 31/435, A61P 11/06
Метки: лечения, бронхиальной, ингибирования, фосфодиэстеразы, способ, 4-триазоло[4, 3-a]пиридины, трициклические, фармацевтическая, 4-с]-1, типа, композиция, астмы, 6-дигидро-9н-пиразоло[3
Код ссылки
<a href="https://by.patents.su/14-4522-triciklicheskie-5-6-digidro-9n-pirazolo3-4-s-1-2-4-triazolo4-3-apiridiny-sposob-ingibirovaniya-fosfodiesterazy-tipa-iv-farmacevticheskaya-kompoziciya-dlya-lecheniya-bronhialnojj-as.html" rel="bookmark" title="База патентов Беларуси">Трициклические 5, 6-дигидро-9Н-пиразоло[3, 4-с]-1, 2, 4-триазоло[4, 3-a]пиридины, способ ингибирования фосфодиэстеразы типа IV, фармацевтическая композиция для лечения бронхиальной астмы.</a>
Производные 5,6-дигидропирона и их фармацевтически приемлемые соли, промежуточные соединения для их получения, фармацевтическая композиция с антивирусной и антибактериальной активностью, способ лечения вызванных ретровирусом инфекции или заболевания

Номер патента: 4282
Опубликовано: 30.03.2002
Авторы: Элизабет ЛАННЕЙ, Эдмунд Ли ЭЛЛСВОРТ, Брадлей Дин ТЕЙТ
МПК: A61K 31/351, A61P 31/12, C07D 309/32...
Метки: получения, приемлемые, активностью, фармацевтическая, промежуточные, производные, фармацевтически, заболевания, лечения, инфекции, вызванных, композиция, антивирусной, 5,6-дигидропирона, способ, ретровирусом, соединения, соли, или, антибактериальной
Текст:
...кислоты. 1. Производные 5,6-дигидропирона по п. 8, где К 3 - группа формулы -Аг 2-СН 23 П 1 п 4-К 12. Производные 5,6-дигидропирона по п. 11, выбранные из группы, включающей 56-дигидро-4 окси-б-(Зметилбутил)-3-2(1-метилэтил)фенилтио 6-фенил 2 Н-пиран-2-он,3-5-этил-2-(1-метил-2-оксиэтил)фенилтио 5,6-ди 1 идро-4 окси-6,6-дифенил-2 Н-пиран-2-он 5-5-(2-циклопентил-5-изопропилфенил)тио-3,6-дигидро 4 окси 6 оксо 2 фенил-2 Нпиран-2-илпентановую...
Соединение, фармацевтическая композиция и способ ингибирования ВИЧ-протеазы

Номер патента: 4552
Опубликовано: 30.06.2002
Авторы: РОДРИГЕС, Майкл Дж., ШЕПЕРД, Тимоти Э., ДЖАНГХАЙМ, Луис Н., КЕЙЛИШ, Винсент Дж., ТЭТЛОК, Джон Х., РАЙЧ, Зигфрид Хайнз, ХЭММОНД, Марлиз, ФРИЦ, Джеймс Э., МАНРОУ, Джон Э., ДРЕССМЕН, Брюс А., КЭЛДОР, Стивен У., ХОРНБЕК, Вильям Дж.
МПК: A61K 31/47, A61K 38/07, A61K 38/02...
Метки: ингибирования, вич-протеазы, способ, композиция, соединение, фармацевтическая
Текст:
...инертен к протекающей реакции и реактанты солюбилизированы в достаточной степени для осуществления желаемой реакции. Типичные растворители для этой реакции включают галогенированные растворители, такие, как метиленхлорид, трихлорэтан, четыреххлористый углерод и т.п. В альтернативном случае альдегид может быть полученс использованием,например, диметоксиметана и подходящей кислоты. В реакции . 3 соединение, выделенное после проведения реакции...
Производные N-(4-пиперидинил) (дигидробензофуран или дигидро-2H-бензопиран)карбоксиамида, способ их получения, фармацевтическая композиция на их основе, промежуточные соединения

Номер патента: 4186
Опубликовано: 30.12.2001
Авторы: ДЕ КЛЕЙН, Михел Анна Йозеф, ВАН ДАЛЕ, Георгес Хенри Паул, БОСМАНС, Ян-Паул Рене Мари Андре
МПК: A61K 31/4523, A61P 1/14, A61K 31/16...
Метки: соединения, производные, фармацевтическая, или, промежуточные, композиция, способ, дигидробензофуран, n-(4-пиперидинил, получения, основе, дигидро-2h-бензопиран)карбоксиамида
Текст:
...гидроксидов, адкоксидов, гидридов, амидов или оксидов (карбонат натрия, гидрокарбонат натрия, карбонат калия, гидроксид натрия, метоксид натрия, гидрид натрия, амид натрия, карбонат кальция, гидроксид кальция, оксид кальция и др.), или неорганические основания, например третичный амин (,-диэтилэтанамин, -/1-метилэтил/-2-пропанамин, 4 этилморфолин и др.), чтобы нужная кислота нейтрализовалась в процессе реакции. В некоторых случаях...
Комбинация атовакуона и прогуанила для лечения протозойных инфекций, способ лечения, фармацевтическая композиция, препарат

Номер патента: 4013
Опубликовано: 30.09.2001
Авторы: Мэри ПАДНИ, Уинстон, Эдвард ГАТТЕРИДЖ, Виктория, Сузан ЛАТТЕР, Дэвид, Брайан, Аштон ХАТЧИСОН
МПК: A61K 31/155, A61K 31/122
Метки: протозойных, фармацевтическая, лечения, комбинация, препарат, прогуанила, атовакуона, инфекций, композиция, способ
Текст:
...удобства помещают в контейнеры для упаковок на один прием и для многократного приема, которые герметизируют после введения препарата и хранят до момента использования. Альтернативно, активные ингредиенты перед применением могут быть в виде порошка, который находится в подходящем носителе,таком как стерильная апирогенная вода. Комбинация атовакуона и прогуанила может быть приготовлена в виде депо-препарата пролонгированного действия, который...
Применение креатинфосфата для ингибирования или снижения роста опухоли и фармацевтическая композиция

Номер патента: 4184
Опубликовано: 30.12.2001
Авторы: ИЗРАЭЛЬ, Виктор, БРЮ, Николь
МПК: A61K 31/66
Метки: или, применение, композиция, ингибирования, снижения, фармацевтическая, опухоли, креатинфосфата, роста
Текст:
...вызывали подкожным инокулированием раковых клеток. Через неделю после инокулирования внутрибрюшной инъекцией вводили креатинфосфат, обозначенный далее как-999-247, или . В процессе лечения животных взвешивали, и размер опухоли измеряли каждую неделю. 1.2. Методика. 1.2.1. Развитие рака человека у голых мышей. Четырехнедельных голых мышей штаммавыращивали в изоляторе (стерильная среда) под давлением. Линии раковых клеток человека, а именно...
Предыдущий патент: Транспортное средство с наклоняемым кузовом
Следующий патент: Короткозамкнутый ротор для асинхронной машины
Случайный патент: Охладитель-аккумулятор естественного холода с водораспылителем