Способ получения порошка нанокристаллического гидроксиапатита
Номер патента: 16625
Опубликовано: 30.12.2012
Авторы: Лесникович Лариса Александровна, Уласевич Светлана Александровна, Крутько Валентина Константиновна, Мусская Ольга Николаевна, Кулак Анатолий Иосифович






Текст
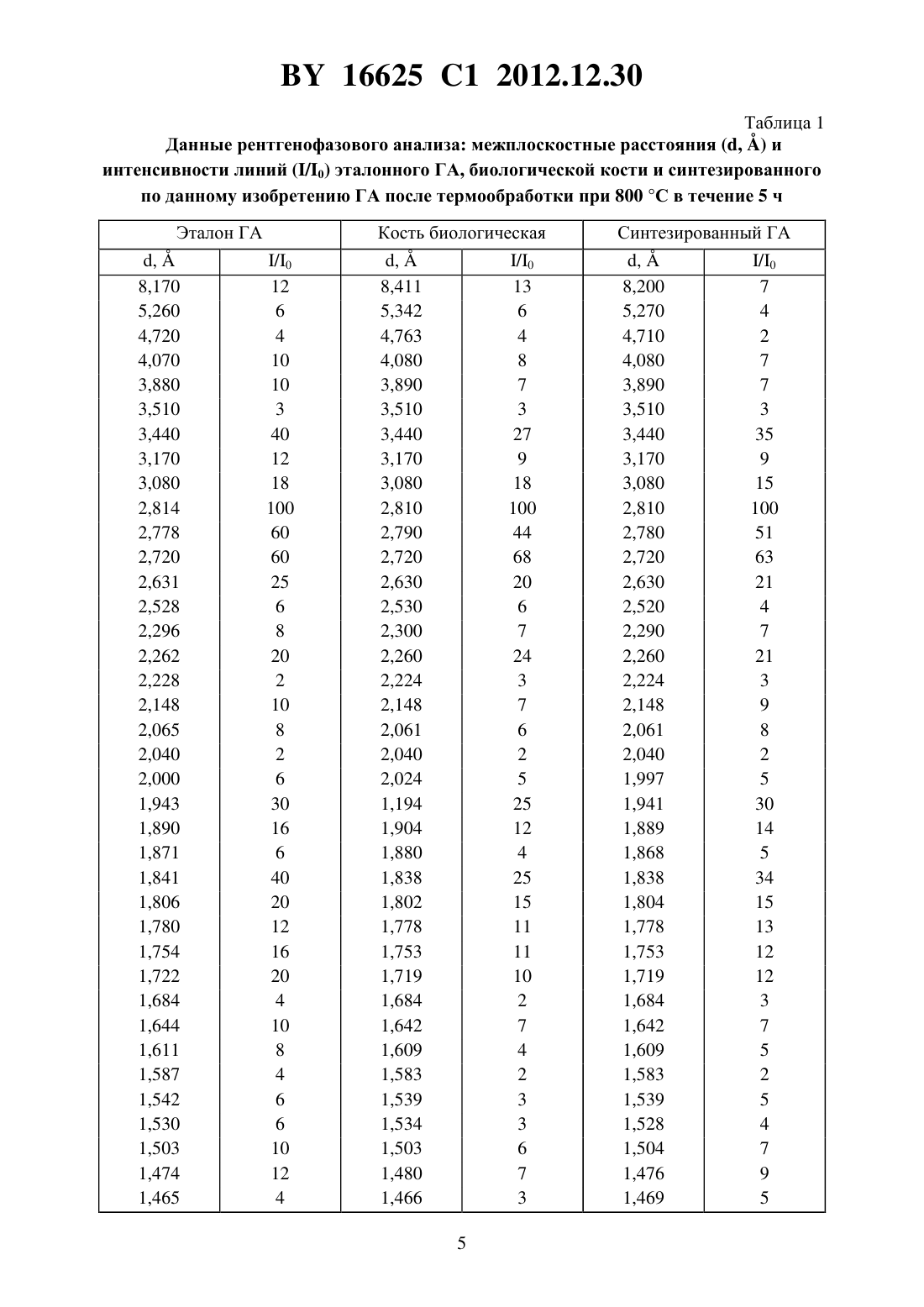
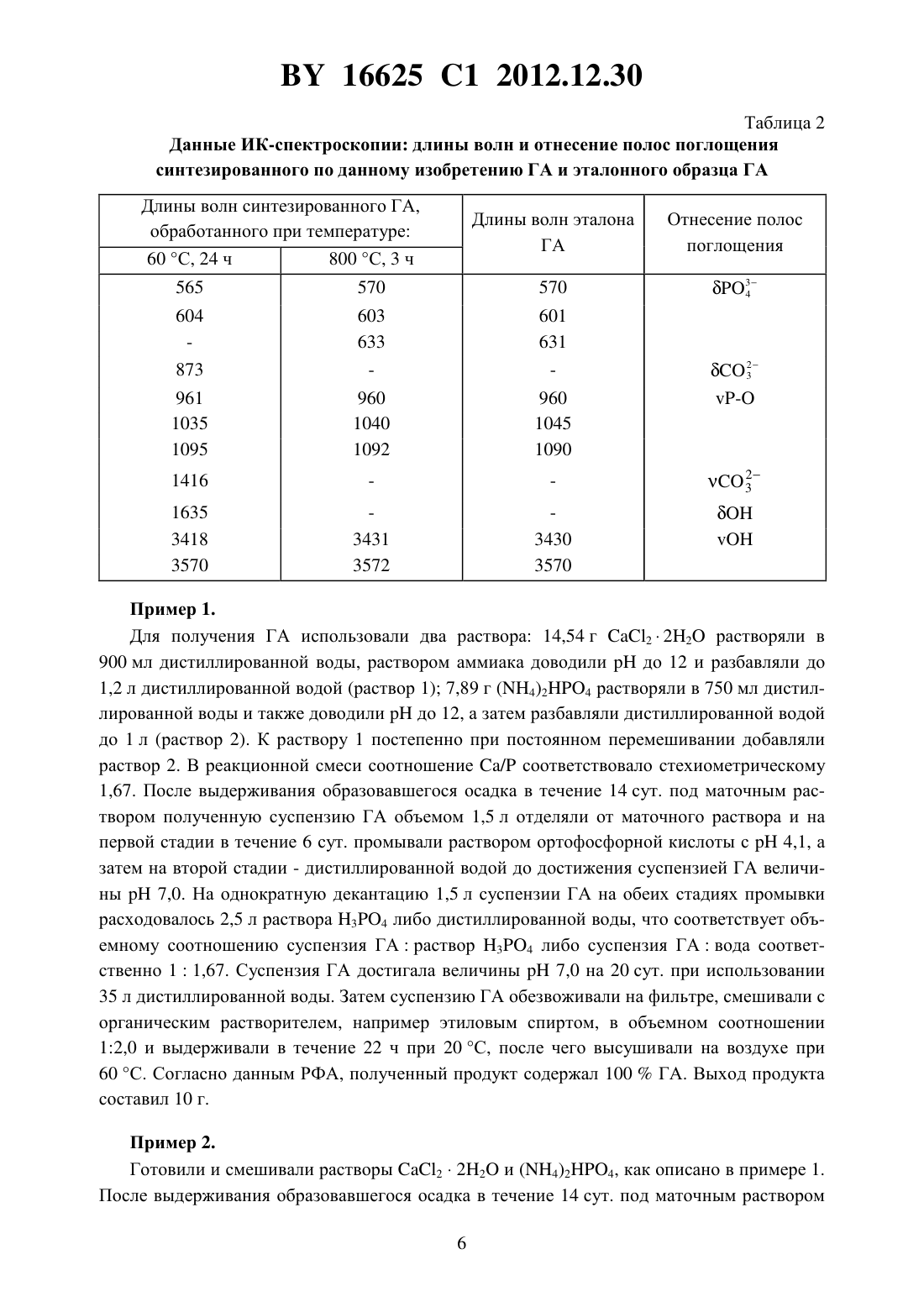
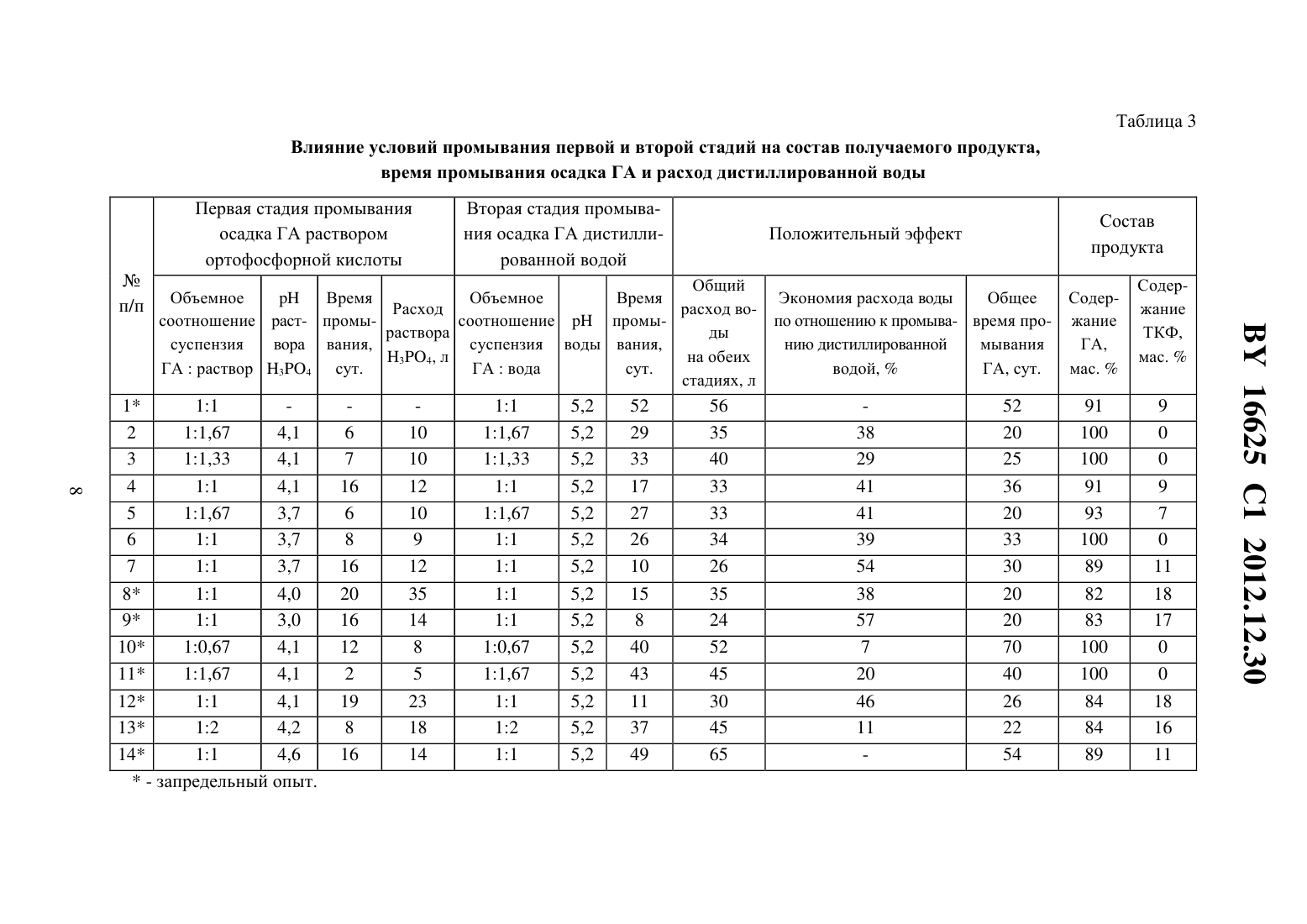
(51) МПК НАЦИОНАЛЬНЫЙ ЦЕНТР ИНТЕЛЛЕКТУАЛЬНОЙ СОБСТВЕННОСТИ СПОСОБ ПОЛУЧЕНИЯ ПОРОШКА НАНОКРИСТАЛЛИЧЕСКОГО ГИДРОКСИАПАТИТА(71) Заявитель Государственное научное учреждение Институт общей и неорганической химии Национальной академии наук Беларуси(72) Авторы Уласевич Светлана Александровна Кулак Анатолий Иосифович Крутько Валентина Константиновна Мусская Ольга Николаевна Лесникович Лариса Александровна(73) Патентообладатель Государственное научное учреждение Институт общей и неорганической химии Национальной академии наук Беларуси(57) Способ получения нанокристаллического гидроксиапатита, включающий взаимодействие водных растворов соли кальция и гидрофосфата аммония с образованием суспензии,отделение осадка и последующую сушку, отличающийся тем, что полученную суспензию выдерживают в течение 1-14 суток, осадок промывают раствором ортофосфорной кислоты с 3,6-4,2 в течение 6-16 суток при объемном соотношении суспензии гидроксиапатита и раствора ортофосфорной кислоты 1(1-1,7), затем промывают дистиллированной водой при объемном соотношении суспензии гидроксиапатита и воды 1(1-1,7) и обезвоживают органическим растворителем. Изобретение относится к способу получения порошка нанокристаллического гидроксиапатита (ГА), используемого для получения биокерамики и композиционных материалов в стоматологии, травматологии, ортопедии и челюстно-лицевой хирургии, а также для нанесения биоактивных покрытий на металлические имплантаты. Известен способ получения порошка нанокристаллического ГА взаимодействием суспензии гидроксида кальция и фосфорной кислоты, взятых при стехиометрическом соотношении 1. В первом реакторе в гомогенную суспензию гидроксида кальция добавляют фосфорную кислоту таким образом, чтобы регулировать щелочное значениев результирующей смеси. Затем данную смесь для ее фазовой трансформации в ГА помещают во второй реактор, где смесь выдерживают при 8-9 в течение 10-20 с, далее смесь загружают в третий реактор и добавляют в реактор суспензию гидроксида кальция с последу 16625 1 2012.12.30 ющей загрузкой образовавшейся смеси снова в первый реактор. Все три стадии повторяются циклично, непрерывно, с определенной периодичностью во времени в конце процесса получается нанокристаллический ГА. Недостатком метода является многостадийность процесса, что значительно усложняет технологию получения ГА, в связи с чем возникает необходимость дополнительных энергетических затрат. Кроме того, для осуществления процесса необходимо специальное оборудование, состоящее из трех реакторов и позволяющее проводить цикличный процесс непрерывно. Полученный продукт может содержать различные примеси, среди которых непрореагировавший гидроксид кальция обладает раздражающим действием и может вызывать воспалительные процессы в костной ране, что не соответствует требованиям гемолитического теста 2. Известен способ получения нанокристаллического ГА осаждением из водных растворов хлорида кальция и фосфата натрия в присутствии гидроксида натрия 3. Для получения ГА к раствору хлорида кальция добавляют раствор 34 /и перемешивают в течение 24 ч. После окончания реакции побочный продукт (хлорид натрия) удаляется периодическим промыванием дистиллированной водой с отделением осадка центрифугированием. Полученная паста высушивается на воздухе. Недостатком данного метода является снижение величиныв процессе синтеза ГА,что приводит к образованию большого количества сопутствующей примеси трикальцийфосфата (ТКФ). Для поддержания определенной величиныв реактор необходимо постоянно вводить гидроксид натрия на протяжении всего времени реакции, что осложняет процесс и увеличивает энергетические затраты. Известен способ получения наноразмерного ГА путем осаждения его в растворе желатина 4. К раствору нитрата кальция добавляется раствор аммиака и раствор желатина, а затем по каплям добавляется гидрофосфат аммония в течение 10 мин при температуре 10 С. Далее смесь перемешивается в течение 2 ч и оставляется на сутки для старения. Значениесреды поддерживается выше 10,5. Через сутки выпавший осадок отфильтровывается и сушится при 120 С. Затем порошок просеивается через сито 60 мкм, дезагрегируется помолом в этаноле, прокаливается при 900 С в течение 1 ч. Недостатком данного метода является необходимость сушки и прокаливания полученного ГА при высоких температурах, что делает процесс не только более энергозатратным,но и способствует снижению биологической активности полученного нанокристаллического ГА 5. Кроме того, термическое разложение желатина, химически связанного с ГА,происходит при 514 С, что может приводить к образованию карбонатзамещенного гидроксиапатита (КГА) 4, который в свою очередь при более высоких температурах прогрева может разлагаться до ТКФ, вследствие чего конечный продукт может быть загрязнен примесями ТКФ и КГА. Наиболее близким к предлагаемому изобретению является способ получения нанокристаллического ГА путем взаимодействия водных растворов сахарата кальция и гидрофосфата аммония, затем образовавшуюся суспензию нагревают до 60 С, отфильтровывают и высушивают в тонком слое. Раствор сахарата готовят взаимодействием прокаленного при 1000 С оксида кальция и раствора сахарозы 6 (прототип). Недостатком данного способа получения ГА является двухстадийность процесса, где вначале прокаленныйреагирует с раствором сахарозы, а затем сахарат кальция взаи 2 16625 1 2012.12.30 модействует с раствором гидрофосфата аммония, что значительно усложняет технологию получения ГА, и продукт может быть загрязнен примесью гидроксида кальция. Кроме того, в данном способе отсутствует стадия промывания, в то время как в предыдущих исследованиях 2 была доказана значимость стадии промывания полученного в щелочной среде ГА. Также при получении ГА выделяется газообразный аммиак, что значительно усложняет процесс и требует специального оборудования для удаления аммиака. Задача, решаемая данным изобретением, заключается в разработке способа получения нанокристаллического ГА с регулируемым фазовым составом в одну стадию. Еще одной задачей настоящего изобретения является оптимизация способа получения нанокристаллического ГА и сокращение времени процесса, уменьшение расхода дистиллированной воды при промывке и, как следствие, уменьшение себестоимости ГА. В соответствии с изобретением описывается способ получения порошка нанокристаллического гидроксиапатита, включающий взаимодействие водных растворов соли кальция и соли фосфорной кислоты при величине 12, выдерживание полученной суспензии под маточным раствором в течение 1-14 сут., промывание полученного продукта раствором ортофосфорной кислоты с 3,6-4,2 в течение 6-16 сут. при объемном соотношении суспензия гидроксиапатитараствор - 1(1-1,7) с последующим промыванием продукта дистиллированной водой при объемном соотношении суспензия гидроксиапатитавода 1(1-1,7), его обезвоживание, обработку органическим растворителем в объемном соотношении 1(1,9-2,1) и сушку на воздухе при 60 С. В предпочтительном варианте реализации изобретения в качестве соли кальция используют хлорид кальция, а в качестве соли фосфорной кислоты используют гидрофосфат аммония при соотношении 23 , равном 1,67. 4 Экспериментально установлено, что использование на первой стадии раствора ортофосфорной кислоты в течение 6-16 сут. позволяет увеличить скорость седиментации полученного ГА (в среднем от 0,004-0,070 см/мин до 0,4-0,8 см/мин) и способствует более быстрому снижению величинысуспензии ГА, чем при промывании ГА дистиллированной водой (примерно в 2-3,5 раза). Однако промывание суспензии ГА до величины 7,0 раствором ортофосфорной кислоты приводит к образованию большого количества ТКФ (свыше 18 ), что не соответствует требованиям ТУ 100049731.090-2006. Поэтому целесообразно использовать комбинированную промывку, где на первой стадии в течение 6-16 сут. промывка осуществляется раствором ортофосфорной кислоты, а на второй стадии - дистиллированной водой. Использование раствора ортофосфорной кислоты на первой стадии обеспечивает более высокую скорость седиментации (0,4-0,8 см/мин) суспензии ГА и снижение величинысуспензии ГА в течение более короткого времени,чем при промывании дистиллированной водой. В частности, при промывке раствором ортофосфорной кислоты с 3,6-3,8 суспензия ГА, рассчитанная на получение 10 г ГА, достигает 9,0-9,2 на 3-6 сут., при промывке раствором ортофосфорной кислоты с 4,04,2 такая же суспензия ГА достигает той же величинына 6-9 сут., в то время как при промывании дистиллированной водой суспензия ГА достигает такой же величинына 11-13 сут. На второй стадии промывания дистиллированной водой ГА сохраняет большую скорость седиментации (0,4-0,8 см/мин), а снижение величинысуспензии ГА в 1,3-2 3 16625 1 2012.12.30 раза быстрее, чем при промывании только дистиллированной водой. Промывка дистиллированной водой на второй стадии необходима, чтобы получить однофазный ГА или двухфазный ГА с регулируемым количеством -ТКФ. Экспериментально показано, что в зависимости от способа промывания раствором ортофосфорной кислоты возможно получение двух различных форм продукта однофазного ГА и двухфазного ГА, содержащего заданное количество -ТКФ (до 11 ). При промывании суспензии ГА в течение 6-14 сут. раствором ортофосфорной кислоты с 3,6-3,8 до достижения величины 7,4-8,5 с последующим промыванием дистиллированной водой получается ГА с регулируемым количеством примеси -ТКФ (до 11 ). При этом промывка протекает в 2-3 раза быстрее, чем при промывании ГА дистиллированной водой, а экономия расхода дистиллированной воды достигает 41-54 . Промывание суспензии ГА в течение 6-8 сут. раствором ортофосфорной кислоты с4,0-4,2 до достижения величины 9,0-9,5 с последующим промыванием дистиллированной водой протекает в 1,5-2 раза быстрее, чем при промывании ГА дистиллированной водой, и при этом получается однофазный ГА, а экономия расхода дистиллированной воды достигает 38-41 . При этом был выявлен эффект, что при использовании на однократную промывку раствора ортофосфорной кислоты либо дистиллированной воды при объемном соотношении суспензия ГАраствор ортофосфорной кислоты (вода), соответственно, 1(1-1,7) происходит сокращение времени промывки в 1,5-3 раза и экономия расхода воды на 20-54 . Промывание суспензии ГА при объемном соотношении суспензия ГАраствор ортофосфорной кислоты меньшим, чем 11 становится достаточно длительным и протекает в 1,5-3 раза дольше, чем при промывании дистиллированной водой. При промывании суспензии ГА при объемном соотношении суспензия ГАраствор ортофосфорной кислоты большим чем 11,7 процесс также сильно растягивается во времени (в 1,3-3 раза дольше,чем при промывании дистиллированной водой) либо продукт получается со значительным количеством -ТКФ (от 16 и выше). Комбинированная промывка с раствором ортофосфорной кислоты с 3,6 нецелесообразна, так как при такой промывке получается ГА с большим количеством -ТКФ(выше 16 ). Комбинированная промывка с раствором ортофосфорной кислоты с 4,4 также нецелесообразна, так как при этом не происходит экономии времени промывания ГА и расхода дистиллированной воды при получении ГА, а в некоторых случаях наблюдается перерасход дистиллированной воды на 18 . По данным рентгенофазового анализа (табл. 1) и ИК-спектроскопии (табл. 2) полученный порошок представлял собой однофазный ГА либо двухфазный ГА с регулируемым количеством -ТКФ (до 11 ). Рентгенограммы эталонного образца ГА 7, неорганической составляющей костной ткани и синтезированного по данному изобретению порошка ГА практически идентичны (табл. 1), что подтверждается и данными ИК-спектроскопии. Методами сканирующей электронной и атомно-силовой микроскопии с послойной самоорганизацией частиц на поверхности кремниевых подложек показано, что полученный ГА представляет собой сферические агломераты размером от 400 нм до 2 мкм, состоящие из наночастиц диаметром от 16 до 30 нм. Согласно критериям 8, такой порошок нанокристаллического ГА является биосовместимым и биоактивным. 16625 1 2012.12.30 Таблица 1 Данные рентгенофазового анализа межплоскостные расстояния (, ) и интенсивности линий (/0) эталонного ГА, биологической кости и синтезированного по данному изобретению ГА после термообработки при 800 С в течение 5 ч Эталон ГА 16625 1 2012.12.30 Таблица 2 Данные ИК-спектроскопии длины волн и отнесение полос поглощения синтезированного по данному изобретению ГА и эталонного образца ГА Длины волн синтезированного ГА,обработанного при температуре 60 С, 24 ч 800 С, 3 ч Длины волн эталона ГА Пример 1. Для получения ГА использовали два раствора 14,54 г 222 растворяли в 900 мл дистиллированной воды, раствором аммиака доводилидо 12 и разбавляли до 1,2 л дистиллированной водой (раствор 1) 7,89 г (4)24 растворяли в 750 мл дистиллированной воды и также доводилидо 12, а затем разбавляли дистиллированной водой до 1 л (раствор 2). К раствору 1 постепенно при постоянном перемешивании добавляли раствор 2. В реакционной смеси соотношение / соответствовало стехиометрическому 1,67. После выдерживания образовавшегося осадка в течение 14 сут. под маточным раствором полученную суспензию ГА объемом 1,5 л отделяли от маточного раствора и на первой стадии в течение 6 сут. промывали раствором ортофосфорной кислоты с 4,1, а затем на второй стадии - дистиллированной водой до достижения суспензией ГА величины 7,0. На однократную декантацию 1,5 л суспензии ГА на обеих стадиях промывки расходовалось 2,5 л раствора 34 либо дистиллированной воды, что соответствует объемному соотношению суспензия ГАраствор 34 либо суспензия ГАвода соответственно 11,67. Суспензия ГА достигала величины 7,0 на 20 сут. при использовании 35 л дистиллированной воды. Затем суспензию ГА обезвоживали на фильтре, смешивали с органическим растворителем, например этиловым спиртом, в объемном соотношении 12,0 и выдерживали в течение 22 ч при 20 С, после чего высушивали на воздухе при 60 С. Согласно данным РФА, полученный продукт содержал 100 ГА. Выход продукта составил 10 г. Пример 2. Готовили и смешивали растворы 222 и (4)24, как описано в примере 1. После выдерживания образовавшегося осадка в течение 14 сут. под маточным раствором 16625 1 2012.12.30 суспензию ГА (1,5 л) отделяли от маточного раствора и промывали раствором ортофосфорной кислоты с 4,0 до достижения суспензией ГА величины 7,0. Стадию промывания дистиллированной водой полностью исключили. На однократную декантацию 1,5 л суспензии ГА расходовалось 1,5 л дистиллированной воды, что соответствует объемному соотношению суспензия ГАраствор соответственно 11. Суспензия ГА достигала величины 7,0 на 33 сут. с использованием 35 л раствора ортофосфорной кислоты. Затем суспензию ГА обезвоживали на фильтре, смешивали с органическим растворителем,например этиловым спиртом, в объемном соотношении 11,9 и выдерживали в течение 22 ч при 20 С, после чего высушивали на воздухе при 60 С. Согласно данным РФА, полученный продукт содержал 18-ТКФ. Выход продукта составил 8,9 г. Пример 3. Готовили и смешивали растворы 222 и (4)24, как описано в примере 1. После выдерживания образовавшегося осадка в течение 14 сут. под маточным раствором суспензию ГА (1,5 л) отделяли от маточного раствора и промывали дистиллированной водой без подкисления до достижения суспензии ГА величины 7,0. На однократную промывку 1,5 л суспензии ГА расходовалось 1,5 л дистиллированной воды, что соответствует объемному соотношению суспензия ГАвода соответственно 11. Суспензия ГА достигала величины 7,0 на 52 сут. с использованием 56 л дистиллированной воды. Затем суспензию ГА обезвоживали на фильтре, смешивали с органическим растворителем,например этиловым спиртом, в объемном соотношении 12,1 и выдерживали в течение 22 ч при 20 С, после чего высушивали на воздухе при 60 С. Согласно данным РФА полученный продукт содержал 9-ТКФ. Выход продукта - 10 г. Последующие варианты комбинированной промывки суспензии ГА представлены в табл. 3. Таким образом, как следует из табл. 3, совокупность и последовательность вышеуказанных операций позволяет в одну стадию получить нанокристаллический ГА. При этом в зависимости от способа комбинированной промывки получается порошок нанокристаллического однофазного ГА либо двухфазного ГА с регулируемым количеством -ТКФ,который соответствует требованиям ТУ 100049731.090-2006, а экономия дистиллированной воды при комбинированной промывке по сравнению с промыванием дистиллированной водой достигает 20-53 , время промывки при этом сокращается в 1,5-3 раза. Таблица 3 Влияние условий промывания первой и второй стадий на состав получаемого продукта,время промывания осадка ГА и расход дистиллированной воды Первая стадия промывания осадка ГА раствором ортофосфорной кислоты Вторая стадия промывания осадка ГА дистиллированной водой Время Объемное Время Экономия расхода воды Общее СодерРасход расход восоотношение раст- промысоотношениепромыпо отношению к промыва- время про- жание раствора ды суспензия вора вания,суспензия воды вания,нию дистиллированной мывания ГА,34, л на обеих ГАраствор 34 сут. ГАвода сут. водой,ГА, сут. мас.стадиях, л 1.7169372, 2007 (аналог). 2. ЛЕСНИКОВИЧ Л.А. и др. Синтез и свойства основного ортофосфата кальция // Вести НАН Беларуси. - 1999. - 1. - С. 15-19. 3.7247288, 2003 (аналог). 4. ФОМИН А.С. и др. Наноразмерный гидроксиапатит, синтезированный осаждением в растворе желатина // Доклады Академии наук. - 2006. - Т. 411. -3. - С. - 348-351 (аналог). 5.13684 1, 2010. 6.2362730 2, 2007 (прототип). 7. ЖАРСКИЙ И.М. и др. Свойства и методы идентификации веществ в неорганической технологии / Мн. Фонд фундаментальных исследований. - 1996. - С. 372. 8. ЦУБЕР В.К. Влияние размера частиц на реакционную способность и биохимическую активность гидроксиапатита // Вести НАН Беларуси. - 2004. -1. - С. 37-40. Национальный центр интеллектуальной собственности. 220034, г. Минск, ул. Козлова, 20.
МПК / Метки
МПК: C01B 25/32
Метки: нанокристаллического, получения, гидроксиапатита, порошка, способ
Код ссылки
<a href="https://by.patents.su/9-16625-sposob-polucheniya-poroshka-nanokristallicheskogo-gidroksiapatita.html" rel="bookmark" title="База патентов Беларуси">Способ получения порошка нанокристаллического гидроксиапатита</a>
Способ получения порошка гидроксиапатита

Номер патента: 2302
Опубликовано: 30.09.1998
Авторы: Лесникович Лариса Александровна, Ильющенко Александр Федорович, Оковитый Вячеслав Александрович, Кулак Анатолий Иосифович, Трофимова Ирина Валериановна, Соболевский Сергей Борисович, Царенков Валерий Минович
МПК: C01B 25/32
Метки: порошка, получения, гидроксиапатита, способ
Текст:
...порошок представляет собой однофазный ГА без посторонних примесей. Операцию старения суспензии ГА проводили для протекания вторичных процессов формирования структуры ГА. Если соотношение Са 2 Р 43-1,9, т.е. превышает стехиометрическое 1,67, время старения уменьшается до 1 суток, поскольку процессы протекают быстрее. Если соотношение меньше стехиометрического - 1,6,то время старения увеличивается до 14 суток, т.к. формирование...
Способ синтеза нанокристаллического порошка лантан-бариевого манганита

Номер патента: 11245
Опубликовано: 30.10.2008
Автор: Труханов Сергей Валентинович
МПК: B82B 3/00, C30B 29/10, C01G 45/00...
Метки: способ, нанокристаллического, лантан-бариевого, манганита, синтеза, порошка
Текст:
...выпаривают при 75-150 С до образования геля на основе полиглицерина, и отжигают гель при 250 С в течение 2 ч. Сущность изобретения заключается в том, что при использовании способа синтеза нанокристаллического порошка лантан-бариевого манганита 0,500,503 достигается однородность химического состава магнитного материала с точкой Кюри в области комнатной температуры вследствие использования глицерина в качестве органической матрицы....
Способ получения гидроксиапатита

Номер патента: 3656
Опубликовано: 30.12.2000
Авторы: Морозов Виктор Егорович, Старовойтов Анатолий Афанасьевич, Шпилевская Лариса Евгеньевна, Морозова Анна Антоновна
МПК: C01B 25/32
Метки: способ, гидроксиапатита, получения
Текст:
...печь, подают инертный газ (гелий) и осуществляют плавный подъем температуры до 500 С. Выдерживают при данной температуре 1-2 часа до получения продукта черного цвета. Затем подачу инертного газа отключают, подают воздух и осуществляют подъем температуры в печи до 800 С. Обжиг полупродукта при заданной температуре проводят до получения гидроксиапатита белого цвета. Печь выключают, охлаждают, продукт выгружают, взвешивают и определяют...
Способ получения сапропелевого порошка для буровых растворов

Номер патента: 5014
Опубликовано: 30.03.2003
Авторы: Зазеркин Геннадий Васильевич, Сенкевич Эдуард Станиславович, Толкачева Татьяна Максимовна, Призенцов Александр Иванович
МПК: C09K 7/00
Метки: буровых, сапропелевого, порошка, получения, растворов, способ
Текст:
...требованиям промышленного производства из-за повышенного расхода материала и мощности. Задача изобретения - получить сапропелевый порошок с влажностью до 10 , не требующий дополнительных модификаций и затрат на получение качественных дисперсий,отвечающих требованиям бурения, ремонта и заполнения скважин. Поставленная задача достигается тем, что сапропелевая крошка, полученная из естественного сапропеля, модифицируется щелочными...
Способ получения порошка лиофилизированного антрациклин гликозида

Номер патента: 822
Опубликовано: 15.08.1995
Авторы: Диего Олдани, Карло Конфалониери, Гаэтано Гатти, Лучиано Гамбини
МПК: A61K 31/715
Метки: способ, гликозида, лиофилизированного, получения, антрациклин, порошка
Текст:
...аминокислоты, такой как, например, цистеин илинапример, фенилаланин или тирозин, или нейтральной гетероциклической аминокислоты, такой как, например, пролин или оксипролин или сочетания двух или более из вышеуказанных соединений.Как известно, некоторые из вышеупомянутых сорастворяюших средств, используются в качестве предохраняющих и баитериостатичесних средств в фармацевтических составах, однако, ничего не известно об их возможной...
Предыдущий патент: Способ частотно-временного преобразования случайного сигнала нелинейного динамического объекта
Следующий патент: Способ поражения низколетящей цели
Случайный патент: Способ нанесения покрытия на цилиндрические детали