N,N’-Замещенные имидодикарбонимидные диамиды,обладающие антипаразитической и антимикробной активностью, профилактическая композиция, композиция для снижения уровня инфекций
Номер патента: 3847
Опубликовано: 30.03.2001
Авторы: Кэнфилд КРЕЙГ, Нейл Дж. ЛЕВИС, Дэвид П. ДЖАКОБУС








Текст
ГОСУДАРСТВЕННЫЙ ПАТЕНТНЫЙ КОМИТЕТ РЕСПУБЛИКИ БЕЛАРУСЬ,-ЗАМЕЩЕННЫЕ ИМИДОДИКАРБОНИМИДНЫЕ ДИАМИДЫ,ОБЛАДАЮЩИЕ АНТИПАРАЗИТИЧЕСКОЙ И АНТИМИКРОБНОЙ АКТИВНОСТЬЮ, ПРОФИЛАКТИЧЕСКАЯ КОМПОЗИЦИЯ, КОМПОЗИЦИЯ ДЛЯ СНИЖЕНИЯ УРОВНЯ ИНФЕКЦИЙ(71) Заявитель ДЖАКОБУС ФАРМАСЬЮТИКАЛ КОМПАНИ, ИНК.(73) Патентообладатель ДЖАКОБУС ФАРМАСЬЮТИКАЛ КОМПАНИ, ИНК.(57) 1. ,-Замещенные имидодикарбонимидные диамиды общей формулы где 1 - С 1-С 6-алкилен, незамещенный или замещенный С 1-С 6-алкилом 3 - С 1-С 6-алкил, незамещенный или замещенный С 1-С 6-алкилом, или фенил, замещенный галогеном 5 - С 1-С 10-алкил, незамещенный или замещенный С 1-С 6-алкилом С 3-С 7-циклоалкил терагидропиранил фенил или бензил, которые могут быть моно- или полизамещены галогеном, нитрогруппой, С 1-С 6-алкилом,С 1-С 6-алкоксигруппой, или нафтил 6 - водород 7 - водород, С 1-С 6-алкил, незамещенный или замещенный С 1-С 6-алкилом, С 1-С 6-алканоил, фенил, бензил- кислород или сераравно 0 или 1 равно 1,или их фармацевтически приемлемые кислотно-аддитивные соли или гидраты указанных солей. 2. Соединение по п. 1, в котором 5 выбран из группы, включающей метил, этил, н-пропил, изопропил, нбутил, изобутил, н-пентил, изопентил и н-децил, незамещенные или замещенные метилом или этилом циклопентил, циклогексил, циклогептил фенил, бензил, фенэтил, фенилпропил, моно- или полизамещенные метилом, этилом или галогеном. 3. Соединение по п. 1, в котором 3 выбран из группы, включающей метил, этил и изопропил. 4. Соединение по п. 1, в котором 5 выбран из группы, включающей метил, этил, н-пропил, изопропил, изобутил, н-пентил, н-децил, циклопентил, циклогексил, циклогептил и фенил. 5. Соединение по п. 1, в котором 5 выбран из группы, включающей С 1-С 10-алкил, замещенный метилом или этилом, фенил или бензил, моно- или полизамещенные метилом, этилом, нитрогруппой, метокси, этокси, пропокси, хлором, бромом или фтором.,где- фенил, - целое число от 1 до 4, - кислород,1 - (2), где- целое число от 1 до 4,3 - изопропил,его таутомеры или нетоксичная кислотно-аддитивная соль. 7. Соединение по п. 1, которое представляет собой -3-(2,4,5-трихлорфенокси)пропокси(1 метилэтил)имидодикарбонимидный диамид или его нетоксичная кислотно-аддитивная соль. 8. Соединение по п. 1, которое представляет собой -3-(2,5-дихлортиофенокси)пропокси(1 метилэтил)имидодикарбонимидный диамид или его нетоксичная кислотно-аддитивная соль. 9. Соединение по п. 1, которое представляет собой -3-(4-хлортиофенокси)пропокси(1 метилэтил)имидодикарбонимидный диамид или его нетоксичная кислотно-аддитивная соль. 10. Соединение по п. 1,которое представляет собой-ацетокси 3-(2,4,5-трихлортиофенокси)пропокси(1-метилэтил)имидодикарбонимидный диамид или его нетоксичная кислотноаддитивная соль. 11. Соединение по п. 1, которое представляет собой -3-(2,4,5-трихлортиофенокси)этокси(1 метилэтил)имидодикарбонимидный диамид или его нетоксичная кислотно-аддитивная соль. 12. Соединение по п. 1, которое представляет собой -(3,4-дихлорбензокси)(1-метилэтил)имидодикарбонимидный диамид или его нетоксичная кислотно-аддитивная соль. 13. Соединение по п. 6, которое представляет собой -3-(2,4,5-трихлорфенокси)пропокси(1 метилэтил)имидодикарбонимидный диамид гидрохлорид моногидрат. 14. Соединение по п. 1, которое представляет собой -ацетил 3-(2,4,5-трихлорфенокси) пропокси-(1-метилэтил)имидодикарбонимидный диамид или его нетоксичная кислотно-аддитивная соль. 15. Соединение по п. 1, которое представляет собой -3-(2,4,5-трихлорфенокси)этокси(1 метилэтил)имидодикарбонимидный диамид или его нетоксичная кислотно-аддитивная соль. 16. Соединение по п. 1, в котором 3 выбран из группы, включающей метил, этил, н-пропил, изопропил,изобутил, н-пентил. 17. Соединение по п. 1, обладающее антипаразитической активностью в отношении. и антимикробной активностью в отношении. и. 18. Соединение по п. 17, обладающее антипаразитической активностью в отношениии антимикробной активностью в отношении М., М.и М. . 19. Профилактическая композиция для защиты от инфекций, вызванных организмом, выбранным из группы, включающей.,. и, субъектов, подверженных вышеуказанным инфекциям, содержащая профилактически эффективное количество соединения по п. 1 и фармацевтически приемлемый носитель. 20. Профилактическая композиция по п. 19, составленная для орального введения. 21. Профилактическая композиция по п. 20, составленная для введения в виде таблеток или капсул. 22. Композиция для снижения уровня инфекций у субъектов, страдающих от инфекций, вызванных организмом, выбранным из группы, включающей.,. и, содержащая эффективное для уменьшения инфекции количество соединения по п. 1 и фармацевтически приемлемый носитель. 23. Композиция по п. 22, составленная для орального введения. 24. Композиция по п. 23, составленная для введения в виде таблеток или капсул. 3847 1 Изобретение относится к -, -замещенным асимметричным имидодикарбонимидным диамидам, получаемым из гидроксиламинов, и к их производным и к способам их получения. Родственные производные триазина (, Е., ... 25659-74) (1989) плохо абсорбируются и, как было показано, менее эффективны, при оральном введении по сравнению с инъекцией, в излечении обезьян , зараженных малярией. Родственные производные триазина приходится вводить с помощью инъекции для получения активности,сравнимой с активностью других известных антималярийных лекарств или превосходящих активность этих лекарств.. 74393-404 (1980).. . . . . . 769-14. , . ., . (1982), , 4.232.022, . 4,179,562). Кроме того, такие триазины, как сообщалось, плохо переносимы при даче их оральным путем (,. ., . (1982) ). ДЕ-А 824942 на имя Империал Кемикал Индастриз раскрывает антималярийные арильные бигуаниды, в которых арильная группа соединена непосредственно с одним из бигуанидных азотов. Предлагаются новые фармацевтически активные соединения формулы,причем все они описаны общей формулой . Любая из этих формул, используемых здесь, должна рассматриваться как эквивалент ее и включает другие. В формуле 1 представляет собой замещенную или незамещенную двухвалентную алифатическую группу с 1-16 атомами углерода, в которой заместители являются моно- или поли- и выбраны из группы, состоящей из низшего алкила, арила или аралкила, 3 3847 1 3 выбран из группы, состоящей из той же группы значений, что и 5, и может также образовывать с азотом, к которому он присоединен, насыщенный гетероцикл с 4-8 атомами углерода,5 выбран из группы, состоящей из замещенного и незамещенного алкила с 1-10 атомами углерода, циклоалкила, гетероциклоалкила с 3-8 атомами углерода, моно- и поликарбоциклоарила с 4-7 атомами в кольце, в котором заместители являются моно- или поли- и выбраны из группы, состоящей из низшего алкила, низшего галоидалкила, циклоалкила с 3-8 атомами углерода, низшего алкенила, низшего алкинила, нитро-, низшего алкокси,низшего алкоксикарбонила, низшего алкилфенила, фенила, моно- и полигалоидфенила, фенокси, моно- и полигалоидфенокси и галогена, при условии, однако, что такое замещение имеет место в моно- и поликарбоциклоариле с 4-7 атомами в кольце,6 и 7 могут быть одинаковыми или различными, когда 6 - водород, алканоил или алкоксиалканоил, и могут также образовывать с азотом, к которому они присоединены, насыщенный гетероцикл с 4-8 атомами углерода,7 может быть также выбран из группы, состоящей из той же группы значений, что и 5, представляет собой кислород или серу, представляет собой 0 или 1, представляет собой 0 или 1 при условии, что, если не указано иное, приставка алк обозначает части молекулы, представляющие собой прямую цепь или разветвленную цепь, и термин низший обозначает 1-6 атомов углерода, а термин алк без приставок обозначает 1-24 атома углерода, их фармацевтически приемлемые соли, аддитивные соли, гидраты указанных солей и аддитивных солей и их моно- и диацилпроизводные соединения. Соединения в объеме настоящего изобретения имеют антимикробную антипаразитическую активность различного рода, включая антималярийную активность, и обеспечивают новую фармакологическую активность, так как, в отличие от вышеупомянутых производных триазина, родительское соединение и его производные, описанные здесь, высоко биодоступны благодаря их способности легко абсорбироваться при оральном введении. Раскрывается способ синтеза новых соединений, согласно настоящему изобретению, путем взаимодействия соответствующим образом замещенного гидроксиламина, тиомина или изостерического амина с замещенным дицианодиамидом в присутствии кислотного катализатора с образованием двузамещенного имидодикарбонимидного диамида с заместителями прии . Эти продукты могут быть затем переведены в соль и далее подвергаться взаимодействию для получения дополнительных заместителей в бигуаниде. Вышеупомянутые замешенные гидроксиламины могут быть синтезированы следующим образом. Вышеприведенный способ осуществим, когдаявляется кислородом или серой и 7 является водородом, алканоилом или алкоксиалканоилом. Однако, когда 7 выбран из группы 5, желателен иной способ получения соединенияи затем .. Также предлагаются способы защиты субъектов (подверженных нижеуказанным инфекциям) от инфекций, вызываемых организмами, выбранными из группы, состоящей из.,., которые предусматривают введение субъекту, подверженному инфекции при контакте 4 3847 1 его с такими организмами, профилактически эффективного количества соединения вышеуказанной формулы. Также предлагаются способы снижения уровня инфекции у субъектов, страдающих от инфекций, вызванных организмом, выбранным из вышеуказанной группы, которые предусматривают введение таким субъектам эффективного количества соединения формулы . Предлагаются также профилактические и лечебные композиции для вышеуказанных целей, которые включают в себя профилактическое или эффективное для снижения инфекции количество соединения формулыи фармацевтически приемлемый носитель. Такие композиции могут быть составлены для орального введения, причем при таком способе введения эти соединения и композиции хорошо абсорбируются, особенно в виде таблеток или капсул. Предлагаются фармацевтически активные соединения формулы,где 1 представляет собой замещенную или незамещенную двухвалентную алифатическую группу с 1-16 атомами углерода, предпочтительно низший алкил, такой, как метил, этил, н-пропил, изопропил, изобутил,н-пентил, н-децил, или циклоалкил, такой, как циклопентил, циклогексил, циклогептил. Заместители суть моно- или поли- и выбраны из группы, состоящей из низшего алкила, такого, как метил, этил, н-пропил, изопропил, изобутил, н-пентил, н-децил, или циклоалкил, такого, как циклопентил, циклогексил, циклогептил,арила, например, фенила, нафтила, тетрагидронафтила, инданила, инденила, бензофуранила и аралкила, такого, как бензил и фенэтил,3 выбран из группы, состоящей из той же группы значений, что и 5, и, если это желательно, он может также образовывать с азотом, к которому он присоединен, насыщенный гетероцикл из 4-8 атомов углерода,такой, как пирролидил, пиперидинил или пирролидинил,5 выбран из группы, состоящей из замещенного или незамещенного алкила с 1-10 атомами углерода, такого, как метил, этил, н-пропил, изопропил, изобутил, н-пентил, н-децил, или циклоалкила, такого, как циклопентил, циклогексил, циклогептил, арила, предпочтительно фенила, нафтила, тетрагидронафтила, инданила, инденила, бензофуранила, бензопиранила, бифенилила, гетероциклоалкила, такого, как тетрагидрофуран,пирролидинил, пиперидил и морфолинил, где заместители суть моно- или поли- и выбраны из группы, состоящей из низшего алкила, такого, как метил, этил, н-пропил, изопропил, изобутил, н-пентил, низшего галоидалкила, такого, как трифторметил, или циклоалкила, такого, как циклопентил, циклогексил или циклогептил, низшего алкенила, такого, как этенил, н-пропенил, изопропенил, изобутенил, н-пентенил, низшего алкинила, такого, как этинил, н-пропинил, изопропинил, изобутинил, н-пентинил, нитро-, низшего алкокси,такого, как метокси, этокси, н-пропокси, изопропокси, изобутокси, н-пентокси, низшего алкоксикарбонила,такого, как формилокси, ацетокси, пропионилокси и бутирилокси, низшего алкилфенила, такого, как бензил,фенила, фенокси, моно- и полигалоидфенила, моно- и полигалоидфенокси, где галоидгруппа представляет собой фтор, хлор или бром, которые могут также служить моно- и полизаместителями для вышеуказанных арильных частей молекулы,6 и 7 могут быть одинаковыми или различными и являются водородом или алканоилом, например формилом, ацетилом, пропионилом и бутирилом. Если нужно, они могут также образовывать с азотом, к которому присоединены, насыщенный гетероцикл с 4-8 атомами углерода, такой, как пирролино, пиперидино или пирролидино, представляет собой кислород или серу, представляет собой 0 или 1, представляет собой 0 или 1,фармацевтически приемлемые соли и аддитивные соли соединения и гидраты указанных солей и аддитивных солей. Также предлагаются моно- и диацилпроизводные этих соединений, например алканоил- или аралканоилпроизводные, такие, как ацетил- и бензоилпроизводные. Соединения формулынастоящего изобретения могут быть синтезированы рядом способов путями, из которых нижеследующие являются наиболее часто применяемыми и предпочтительными. В этом многостадийном способе некоторые промежуточные соединения могут быть коммерчески доступны, однако для полноты описания нижеследующее описание способа начинается с исходных материалов, которые легко можно приобрести. В тех случаях, когда цель состоит в получении соединения, гдепредставляет собой кислород или серу иравно 1, исходным материалом является алканол, фенол или меркаптан . Когда в качестве исходного 5 3847 1 материала применяют алканол, берется избыток алканола, и количество его, необходимое для реакции, обрабатывают одним эквивалентом раствора щелочного металла для образования соли щелочного металла в алканольном растворе. В случае меркаптанов или фенолов используется избыток водного раствора щелочи, например гидроксида натрия, которая образует соответствующую натриевую соль при температуре окружающей среды в течение нескольких минут. Затем добавляют избыток, предпочтительно 2-кратный избыток дигалоидалкана по отношению к рассчитанному количеству соли щелочного металла, причем положение галоидных групп определяется длиной части молекулы, обозначаемой 1. Смесь кипятят с обратным холодильником в течение приблизительно от 1 до 4 часов. Добавляют еще избыток щелочи и реакционную смесь поддерживают при температуре от 50 до 70 С в течение приблизительно 1/2 часа. Смесь охлаждают, нижний органический слой отделяют, промывают и перегоняют под пониженным давлением, получая воду, непрореагировавший дигалоидалкан и целевой 5 окси- или тиоалкилгалоид . Ацетогидроксамовую кислоту превращают в соответствующий гидроксамат щелочного металладобавлением алканольного, предпочтительно этанольного, раствора гидроксида щелочного металла, такого, как гидроксид натрия или калия. Затем прибавляют окси- или тиоалкилгалоид , полученный, как описано выше, и смесь кипятят с обратным холодильником, предпочтительно в течение от приблизительно 4 до приблизительно 8 часов, и охлаждают. Осажденную соль галоид щелочного металла удаляют фильтрацией, растворители удаляют под пониженным давлением, а остаток растворяют в полярном, смешивающемся с водой, органическом растворителе, предпочтительно в растворе ацетона, снова фильтруют и концентрируют под пониженным давлением, получая в результате соответствующий окси- или тиоалкилацетогидроксамат . Когдаравно 0, например, когда 5-1 представляет собой бензил, то соответствующее 5-1 галоид соединениеможет быть получено в промышленности, такое, как бензилбромид, которое вводят в реакцию непосредственно с ацетогидроксаматом, как описано выше. Ацетогидроксаматрастворяют в алканоле, к которому добавляют избыток разбавленной минеральной кислоты, предпочтительно хлористоводородной кислоты, смесь кипятят с обратным холодильником в течение приблизительно от 2 до приблизительно 6, предпочтительно от 4 часов, растворители удаляют под пониженным давлением и остаток экстрагируют сухим диэтиловым эфиром. Растворитель затем удаляют под пониженным давлением, а остаток перекристаллизовывают из алканола, предпочтительно этанола или изопропанола, получая заданный алкилоксиамин гидрохлорид . Алкилоксиамингидрохлоридрастворяют в алканоле и обрабатывают концентрированной водной хлористоводородной кислотой до тех пор, пока раствор не станет определенно кислым. Добавляют в избытке соответствующий омега-замещенный дициандиамид, например низший алкилдициандиамид . Смесь кипятят с обратным холодильником в течение от приблизительно 2 до приблизительно 6 часов, растворители удаляют выпариванием под пониженным давлением с получением желаемого гидрохлорида алкокси омегазамещенного иминодикарбонимидного диамида . Это масло, после обработки и растирания с безводным эфиром, дает твердый осадок, который может быть перекристаллизован, предпочтительно из этилацетата,как гидрат. Когда реагентпредставляет собой моно-омега-замещенный дициандиамид, не несущий ни одного заместителя на остающемся иминоазоте, тогда 7 в соединенииявляется водородом, и таким образом полученный продукт формулыне будет иметь заместителей при азотах 2 и 4, то есть 6 и 7 будут водородом. Когда оба азота иминогрупп замещены, тогда 7 будет иным, нежели водород. Когда нужно либо ввести одинаковые заместители у азотов 2 и 4, либо, если 7 отличен от водорода,ввести иной заместитель у азота 2, гидрохлоридгидратсуспендируют в подходящем органическом растворителе, не смешивающемся с водой и инертном для реакции, предпочтительно в этилацетате, встряхивают с избытком водного раствора щелочи, предпочтительно водным раствором гидрокси натрия, отделяют органический слой, сушат и нагревают с обратным холодильником в течение от приблизительно 1 до приблизительно 4 часов с избытком подходящего ацилирующего агента, например ацетилхлоридом. После окончания реакции летучие компоненты удаляют под пониженным давлением, получая заданное 2 - ацилированное соединение. Как показано выше, когда 7 имеет значение, выбранное из группы 5, желателен иной путь синтеза. О методологии см. , . С. . 1630-45 (1948), . .,,1977-8 (1965). К суспензии цианамида натрия в алканоле, например этаноле, прибавляют 3 изотиоцианат , получая осадок натриевой соли -циано-1-3-тиомочевины , который отфильтровывают, промывают алканолом. Добавляют при быстром перемешивании при температуре окружающей среды метилиодид. Продукт разделяют. Суспензию охлаждают на ледяной бане, твердые вещества отфильтровывают и промывают водой и сушат, получая -циано-1-3 метилизотиомочевину . Изотиомочевинуприбавляют к алканольному раствору 7 амина и смесь нагревают в течение 4 часов в толстостенной склянке при температуре приблизительно 50 С. Получающийся в результате прозрачный раствор постепенно разбавляют водой (75 см 3) и продукт кристаллизуется, давая дициано 3, 7 6 3847 1 диамид . Последний может быть затем введен в реакцию с гидроксиламин-гидрохлоридной солью, как описано ранее, с получением желаемого соединения . Соединения, согласно настоящему изобретению, могут быть получены в форме моногидрогалоидных кислотно-аддитивных солей и/или сольватированного соединения, например, гидрохлоридгидрата или гидробромида. Однако другие соли могут быть получены простой реакцией основания с кислотой и могут быть полезны для модификации свойств продукта, таких, как его токсичность, вкус, физическая форма или скорость выделения в тело. Например, соединения могут быть получены в форме пикрата, сахарината, ацетата,кислого малеата, кислого фталата, сукцината, фосфата, нитро-бензоата, стеарата, манделата, ацетилглицината, памоата, сульфоната, дисульфоната, циклогексилсульфамата, цитрата, тартрата или глюконата. Устойчивые соли обычно получаются при соотношении одна молекула ,-полизамещенных имидодикарбонимидных диамидов на 1 или 2 молекулы одноосновной кислоты (или более чем одна молекула соединения 1 в случае многоосновных кислот), но возможность иметь основные группы в качестве заместителей в 5, например, означает, что в некоторых случаях могут быть соединены с двузамещенным имидодикарбонимидным диамидом и большие количества кислоты. Кроме того, вышеуказанные молекулы могут содержать различные гидратированные формы с молекулами воды или другим растворителем, включенным в молекулярную формулу устойчивой структурной единицы. Присутствие иминобигуанидных азотов в молекуле создает возможность образования ацилпроизводных путем реакции с подходящими субстратами. Раскрывается усовершенствованный способ профилактики и лечения инфекций, вызванных одним или более чем одним плазмодием, микобактериями, токсоплазмозными и пневмоцистозными организмами, и агентами, вызывающими нокардиозные инфекции. ,-замещенные асимметричные бигуаниды формулы ,согласно настоящему изобретению и/или их соли и/или производные имеют противомалярийную и противобактериальную активность, а также эффективны против некоторых грибков, простейших, паразитов и вирусов. Кроме того,и -замещенные производные формулыпроявляют подобную же активность. В частности, эти ,-замещенные асимметричные бигуаниды и соли, а также ихи -замещенные производные проявляют активность против паразитов, включая плазмодии малярии, . , проявляют противомикробную активность против микобактерий, включая, но не ограничиваясь этим, ., .,. , .и пневмоцистозные организмы, такие, как . , связанные (но не ограниченные ими) с пациентами с ослабленной иммунной системой. Кроме того, эти соединения имеют активность против нокардиозных инфекций. Эти соединения имеют активность против нокардтозных инфекций. Эти соединения могут также быть потенциированы в комбинации с сульфонамидами или сульфонами для улучшения биологического спектра и возможностей этих соединений формулы . Наши данные об использовании подтверждены дополнительными обширными исследованиями, проведенными на животных, при поддержке Министерства обороны США. Мы нашли, что новые соединения, согласно настоящему изобретению, показывают высокие уровни эффективности при оральном введении по сравнению с родственными производными триазина, о которых известно, что они плохо адсорбируются. В отличие от родственных производных триазина, этот новый ряд соединений не требует введения с помощью инъекции для проявления активности, сравнимой с другими известными противомалярийными лекарствами или превосходящей активность указанных лекарств. Примеры биологической активности изобретения. Биологическая активность против. Метод испытания активности против малярийных паразитов человека описан подробно в . . ,. . . ., 1978, 27718-737. Подробные методы включают все аспекты лечения животных, инфекции и оценки эффективности лекарств. Испытание проводится с помощью скринингав системе, принятой как стандарт для идентификации эффективных антималярийных соединений в человеке. Тест-система использует ночных обезьян (,), родина которых Колумбия. Обезьян инфицируют различными отобранными штаммами малярии посредством внутривенной инокуляции 5106 трофозоитов. Эти трофозоиты получают непосредственно из инфекций ., выделенных из человека, и инфекционные организмы хорошо охарактеризовывают в отношении их ответа на лекарственную терапию. Системауникальна в том, что она делает возможным оценку малярии человека, вызванную . Лекарства вводятся обезьянам через желудочную трубку, и обычная схема исследования включает дачу ежедневных доз подопытным животным в течение семи дней. Активностьопределяют по очищению или уничтожению малярийной инфекции. В таблице 1 (табл. 1-8 см. в конце описания) активность титульного соединения 7776, -3-(2,4,5 трихлорофенокси)пропокси(1-метилэтил)имидодикарбонимидного диамида, сравнивается с двумя известными антималярийными лекарствами и приводятся результаты сравнительных испытаний в весьма стойком к лекарствам Вьетнамском Смитовском штамме, 7776 вызывал четкий ответ на дозу у 8/8 животных, которым вводили 3,0 мг/кг в день в течение трех дней, что указывает на очищение от паразитов (100 -ный ответ). Три из восьми подопытных животных были излечимы (37,5 ). Более 7 3847 1 высокие дозы приводили к более высокому проценту излечения, составлявшему 75 и 100 соответственно при дозах 30,0 и 150,0 мг/кг. Сравнение с прогуанилом и циклогуанилом до 150 мг/кг в течение трех дней показало отсутствие активности (0 ответ). Активность прогуанила против инфекций. Проведены сравнительные испытанияна мышах против . Подтверждающие испытания, проведенные под эгидой Министерства обороны США, демонстрируют благоприятную оральную активность. Результаты демонстрируют превосходную биологическую доступность и эффективность 7776 при оральном способе введения по сравнению с соответствующим ему триазином 99210 и противомалярийным прогуанилом. Эти данные в таблице 2 показывают количество излечений и эффективную дозу, излечивающую 50 зараженных животных (-50), когда лекарства вводились в арахисовом масле подкожно или когда вводились в качестве однократной оральной дозы (РО). Преждевременные смерти животных(раньше, чем через пять дней посте заражения) рассматриваются как указания на токсичность. Таблица 2 приводит данные о пониженной токсичности 7776 в этом скрининг-тесте и превосходную оральную эффективность. Также представлен (в таблице 3) второй широко признанный стандартный тест, демонстрирующий непосредственное сравнение подкожногои орального (РО) введения доз .мышам. Эти тестсистемы подробно описаны в публикациях .. . , 9 . . . . . (1973) 1281 (406)(2) . . , . .. , . . . 1967, 10431. Согласно этим методам, группы в 5 или 10 мышей инфицируют стандартным инокулятом индуцированной кровью инфекции .и лечат однократной подкожной дозой (9 нг/кг) испытуемого лекарства,суспендированного в арахисовом масле, или однократной оральной дозой испытуемого лекарства, суспендированного в гексаметилцеллюлозе и Твеен . Животные затем наблюдаются в течение максимум тридцати дней. Контрольные животные обычно живут 6-7 дней. Чтобы лекарство считалось эффективным, подопытные животные должны прожить по меньшей мере вдвое дольше, чем нелеченные зараженные контрольные животные. Животные, которые проживут тридцать дней, рассматриваются как излеченные. Таблица 4 представляет сравнительные данные по эффективности 7776 против различных штаммов малярии, испытанногос сульфонамидом или без него для определения преимуществ, если таковые есть, такого совместного введения с соединениями, являющимися предметом настоящего изобретения. Результаты, показанные ниже, измеренныев качестве дозы для ингибирования 50 роста (-50) малярийных паразитов в стандартной культуре (. .. . , . . . 1977, 20 237243), представлены в нанограммах на миллилитр (нг/мл). Эти данные показывают, что внутренне присущая активность 7776 возрастает от 4 до 19 раз (см. значения -50) благодаря сульфонамидам в присутствии определенных паразитов, устойчивых к лекарству. Биологическая активность против. Оценка лекарств по их активности противпроводится по широко признанной и хорошо определенной испытательной системе, разработанной и опубликованной Др. Уолтером Т. Хью ( Т. ). На нее широко ссылаются, и она представляет собой общепринятый метод, четко определенный в литературе относительно содержания животных, инфекций, протокола лечения и оценки эффективности по вскрытию и выживаемости. Описание методологии, описанной У. Хью и др. можно найти в .. 1988, 32623-625. Согласно этому методу, иммунную систему крыс подавляли большими дозами глюкокортикостероидов,одновременно защищая их от бактериальной инфекции введением антибиотического тетрациклина. Стандартный метод оценки предусматривает подавление иммунной системы животных стероидами и введение различных доз испытуемых соединений в течение шести недель, за это время у незащищенных животных разовьется пневмоцистозная первичная ацилтическая пневмония. Процент незаболевших животных представляет собой эффективность выбранной дозы испытуемого лекарства. Когда животных подвергают иммуносуспрессии и лечению в соответствии с принятой методологией,обычно наблюдают, что у 75 или более испытуемых животных спонтанно развивается пневмоцистоз. Обычный метод вызвать пневмоцистоз у животных предусматривает введение 2 мг дексаметазона и 50 мг тетрациклина гидрохлорида на литр питьевой воды. Испытуемые соединения подмешиваются в пищу. Положительное лечение обеспечивается контрольным соединением сульфаметоксазол-триметоприм (/),которое вполне эффективно защищает животных от пневмоцистоза при дозах в 250 мг/кгв комбинации с 50 мг/кг ТМР. Другим широко применяемым эффективным соединением является Дапсон при дозе 125 мг/кг . Таблица 5 демонстрирует эффективность 7776 в сравнении с теми известными активными методами лечения, которые применяются для лечения и предупреждения пневмоцистозных инфекций у человека. 7776 эффективен на 100 и также эффективен как Дапсон, который является рекомендуемым антипневмоцистозным лекарством для человека. Активность против микобактериальных инфекций. 8 3847 1 Испытание новых лекарств на активность против микобактериальных инфекций проводитсяисогласно хорошо разработанным лабораторным процедурам, которые широко опубликованы. Методы,используемые для испытания биологической активности против растущего,(МТВ)описаны Гонзалезом и др. . ... . . 1989, 2419-22 .. . . . ., -4,1992, . Активностьопределялась против клинических изолятов , МТВ и МК с использованием метода разбавления бульона. Микобактерии выращивались в течение нескольких дней в 7 Н 10 бульона, рН 6,6,с 10 -ным обогащением пои 0,05 Твин. 80. Последовательные двукратные разбавления антимикробных лекарств готовились в 7 Н 10 бульоне при 128 мкг/мл и менее. Культуры, содержащие окончательную концентрацию приблизительно от 2,5104 до 6,3105 КОЕ/мл, инкубировали на вращающемся встряхивателе при 37 С в течение 7 дней и снимались показания, при этом минимальная ингибирующая концентрация определялась как МС при самой низкой концентрации без видимого помутнения. 7776 в этих опытах сравнивали с известными активными антимикробными лекарствами Прогуанилом , Циклогуанилом ,Сульфаметазиноми/или Дапсоном . Результаты рассматриваются как благоприятные при концентрациях ниже 64 мкг/мл и показаны в таблице 6. 7776 показал превосходство над другими лекарствами. Фармацевтические композиции. Настоящее изобретение предлагает также фармацевтические композиции, содержащие в качестве активного ингредиента соединение, согласно настоящему изобретению, вместе с фармацевтически приемлемым носителем. Водорастворимость гидрохлорида родительского соединения и большинства других солей не очень велика, так что, когда требуются растворы, часто приходится добавлять солюбилизирующие агенты в воду, выбирать неводные растворители или находить более растворимую соль или готовить очень разбавленные растворы. Предпочтительны составы для орального введения, и настоящее изобретение имеет то преимущество над родственными продуктами, что соединения, предлагаемые им, легко поглощаются млекопитающими на достаточном уровне, что делает соединения, согласно настоящему изобретению, орально активными в качестве терапевтических агентов. Составы для инъекций или орального применения основаны на достаточной растворимости, чтобы позволить терапевтическому агенту войти в раствор в желудке или во вспрыскиваемой среде. Лекарственные формы включают таблетки, пилюли, капсулы, мешочки, гранулы, порошки, жевательные резинки, суспензии, эмульсии и растворы, особенно предпочтительны для орального применения таблетки и капсулы во всех вариантах и стерильные растворы для инъекции или инфузии. Там, где это уместно или необходимо, составы могут включать разбавители, связующие агенты, диспергирующие агенты, поверхностно-активные вещества, связывающие агенты, покрывающие материалы, ароматизирующие агенты, красящие агенты, составы с регулируемым выделением активного вещества, подсластители или любые другие фармацевтически приемлемые добавки, например желатин, крахмальный глюколат натрия, лактозу, крахмал,тальк, стеарат магния, микрокристаллическую целлюлозу, Повидон, гидрогенизованные или ненасыщенные масла, полигликоли, сиропы или другие водные растворы. В случае таблеток или капсул и т.п. композиции могут быть представлены в виде предварительно отмеренных единичных доз или в упаковках лекарственных доз для многократного приема, из которых может отбираться подходящая единичная доза. Формой, применимой для инъекций, может быть водный или неводный раствор, суспензия или эмульсия в фармацевтически приемлемой жидкости, например стериальной апирогенной воде или парентерально приемлемых маслах или смеси жидкостей, которые могут содержать бактериостатические агенты, антиоксиданты или другие консерванты и стабилизаторы, буферные смеси (предпочтительно, но не ограничиваясь этим,в физиологических пределах рН от 6,5 до 7,7), растворенные вещества для придания раствору изотоничности с кровью, загустители, суспендирующие агенты или другие фармацевтически приемлемые добавки. Такие формы будут представлены в виде единичной дозы, такой, как ампулы или одноразовые устройства для инъекций, или в виде форм для многократных доз, таких, как бутылочка, из которой может отбираться подходящая доза, или в твердом виде, или в виде концентрата, который может быть использован для быстрого приготовления композиции для инъекции. Все составы для инъекции предпочтительно стерильны и апирогенны. Суппозитории, содержащие соединение, также содержат подходящие носители, например масло какао-бобов, полигликоли или другие носители, известные в современной практике. Помимо стандартных фармацевтических добавок, в рецептуру соединения могут быть включены другие терапевтические агенты, в частности другие антималярийные средства и дезинфектанты. Предпочтительный диапазон дозировок составляет от 0,5 до 10 мг/кг/день. Диапазон довольно обширен,потому что врач сам должен решать, является ли дозировка профилактической и, если она введена инфицированному субъекту, каков уровень инфекции. Если лекарство дается в таблетках, то таблетки могут содержать от 25 до 250 мг активного материала.-3-(2,4,5-трихлорфенокси)пропокси(1-метилэтил)имидодикабонимидный диамид гидрохлорид . Смесь 39,5 г (0,20 моля) 2,4,5-трихлорфенола и 33 мл 25 -ного водного раствора гидроксида натрия объединяют и перемешивают при температуре окружающей среды в течение 15 минут, в течение которых добавляют 80 г (40,7 мл, 0,4 моля) 1,3-дибромпропана. Реакционную смесь кипятят с обратным холодильником в течение 2 часов, в течение которых добавляют дополнительно 51 мл 14-процентного водного раствора гидроксида натрия, и реакционную смесь поддерживают при 50-70 С в течение 30 минут. После охлаждения нижний слой отделяют и промывают пять раз водой. Остаточный органический слой перегоняют при 1 мм рт. ст., получая различные фракции - дистилляционную воду и дибромпропан при 30-40 С и продукт, который перегоняют в температурном интервале от 120 до 157 С. Было собрано 5 г бесцветного масла, которое затвердевает при стоянии, давая 70 -ный выход 3-(2,4,5-трихлорфенокси) пропилбромида . Ацетогидроксамовую кислоту (8,5 г, 0,13 моля) прибавляют к 110 мл этанольного раствора гидроксида натрия (4,0 г, 0,1 моля). Прибавляют 3-(2,4,5-трихлорфенокси)пропилбромида(31,8 г, 0,1 моля) и смесь кипятят с обратным холодильником в течение 6 часов и охлаждают до комнатной температуры. Раствор фильтруют и выпаривают, остаток растворяют в 100 мл ацетона и раствор фильтруют и концентрируют, получая 16,0 г (51 ) 3-(2,4,5-трихлорфенокси)пропилацетогидроксамата , т. пл. 102-104 С. Ацетогидроксамат(31,3 г, 0,1 моля) растворяют в 120 мл метанола. Прибавляют хлористоводородную кислоту (30 мл 12 -ного раствора) и смесь кипятят с обратным холодильником в течение 4 часов. Остаток выпаривают досуха под вакуумом, промывают сухим диэтиловым эфиром и перекристаллизовывают из изопропилового спирта (90 мл), получая (15,5 г, 58,7 ) 3-(2,4,5-трихлорфенокси)пропилоксиамина гидрохлоридам , т.пл. 158-168 С. Гидроксиламин гидрохлорид(10 г, 0,0267 моля) в 160 мл этанола обрабатывают 6 н. водной НС,пока раствор не становился кислым. Прибавляют изопропил-дипиано-диамид (4,4 г, 0,0347 моля) и смесь кипятят с обратным холодильником в течение 4 часов, в течение которых растворитель испаряется. Полученный твердый материал растворяют в воде и этилацетате и образовавшееся масло обрабатывают безводным эфиром, получая твердый осадок, который отфильтровывают, промывают эфиром и сушат. Полученное в результате твердое вещество, перекристаллизованное из этилацетата, после очистки на активированном угле дает 2,0 г титульного соединенияв виде моногидрата с т. пл. 100 С. В соответствии с вышеприведенной процедурой, но там, где вместо 1,3-дибромпропана используют метилендибромид, 1,2-дибромэтан, 1,4-дибромбутан или 1,5-дибромпентан, получают соответственно метокси, этокси-, бутокси- или пентокси-аналог. В соответствии с вышеприведенной процедурой, но там, где вместо 1,3-дибромпропана используют 1,2 дибромпропан, 1,3-дибром-2-метоксипропан, 1,4-дибром-2-этоксибутан или 1,5-дибром-3-этоксипентан, получают соответственно 2-метилэтиловый, 2-метоксипропокси-, 2-этоксибутокси- или 3-этоксипентоксианалог. Пример 2.. Подобно синтезу 2,5-дихлортиофенол (35,8 г, 0,2 моля) обрабатывают гидроксидом натрия (40 мл, 20 ного водного раствора) и затем объединяют с 1,3-дибромпропаном (160 г, 0,8 моля) и кипятят с обратным холодильником в течение 4 часов. Смесь охлаждают, водный слой отделяют и нейтрализуют 20 -ным раствором гидроксида натрия, а нижний слой промывают пять раз водой и перегоняют при 1 мм рт. ст. Основную фракцию собирают в интервале температур от 130 до 145 С в виде бесцветного масла (50 г, 84 ) 2,5 дихлортиофеноксипропилбромида , который подвергают дальнейшему взаимодействию с ацетогидроксамовой кислотой, как описано ранее в примере 1, и гидролизуют для получения 3-(2,5 дихлортиофенокси)пропилоксиамина гидрохлорида , который затем подвергают реакции с изопропилдицианодиамидом, как описано ранее в примере 1, получая титульное соединение . В соответствии с вышеприведенной методикой, но там, где вместо 2,5-дихлортиофенола используют нпропилмеркаптан, циклогексилмеркаптан и 3-тетрагидропиранол, получают соответствующий -3-(-ппилтио-,циклогексилтио- и -3-тетрагидропиранилокси)пропилокси(1-метилэтил)имидодикарбонимидный диамид гидрохлорид. Пример 3.-3-(4-хлортиофенокси)пропилокси(1-метилэтил)имидодикарбонидный диамид гидрохлорид . Подобно синтезу соединения 4-хлортиофенол (28,9 г, 0,2 моля) обрабатывают гидроксидом натрия(40 мл 20 -ного водного раствора) и затем объединяют с 1,3-дибромпропаном (160 г, 0,8 моля) и кипятят с обратным холодильником в течение 4 часов. Смесь охлаждают, водный слой отделяют и нейтрализуют 20 ным раствором гидроксида натрия, а нижний слой промывают пять раз водой и перегоняют при 1 мм рт. ст. Основную фракцию собирают в температурном интервале от 120 до 130 С в виде бесцветного масла (47,5 г,90 ), которое кристаллизуется при стоянии, образуя 4-хлортиофеноксипропилбромид , который затем подвергают дальнейшему взаимодействию с ацетогидроксамовой кислотой, как описано ранее в примере 1, и гидролизуют с 10 3847 1 получением 3-(4-хлортиофенокси)пропилоксиамина гидрохлорида . Последний в свою очередь вводят в реакцию с изопропилдицианодиамидом, как описано в примере 1, с получением титульного соединения . В соответствии с вышеприведенной процедурой, но там, где вместо изопропилдицианодиамида используют-фенилизопропилдицианодиамид или другой -заместитель, такой, как метил, этил или фенилметил, получают соответствующий -3-(4-хлортиофенокси)пропокси -фенил или метил, этил или фенилэтил, (1 метилэтил)имидодикарбоимидный диамид гидрохлорид. Если желательно получить ,-диалканоил или соответствующие моноалканоильные производные вышеуказанных ненасыщенных производных в формуле , последние обрабатывают таким же образом, как указано в примере 4 ниже, с тем чтобы соответствующее молярное соотношение 11 хлорангидрида кислоты или ангидрида для монозамещенных или молярное соотношение 21 для дизамещенных производных обеспечило бы получение продукта. Пример 4.-2-(2,4,5-трихлорфенокси)пропокси(1-метилэтил)имидодикарбонимидный диамид гидрохлорид гидрат(1,0 г, 0,002 моля) суспендируют в этилацетате (20 мл) и встряхивают с 0,1 мл 25 -ного водного раствора гидроксида натрия. Органический слой отделяют и сушат (сульфат магния), добавляют 0,1 мл ацетилхлорида и смесь нагревают в течение 2 часов. В последующем смесь концентрируют с получением 0,5 г (47 ) титульного соединенияв виде белых кристаллов, т. пл. 160-170 С. Пример 5.. Смесь 39,5 г (0,20 моля) 2,4,5-трихлорфенола растворяют в 40 мл 20 -ного водного раствора гидроксида натрия и по каплям прибавляют в течение 1 часа к дибромэтану (85,8 мл, 1 моль), нагреваемому с обратным холодильником. Смесь кипятят с обратным холодильником 2 часа и оставляют охлаждаться до комнатной температуры. После охлаждения нижний слой отделяют и промывают четыре раза водой. Оставшийся органический слой перегоняют при 1 мм рт. ст. с получением основной фракции в температурном интервале от 145 до 155 С в виде бесцветного масла (51,4 г, 85 ), которое было 2-(2,4,5-трихлорфенокси)-этилбромид,Трихлорфеноксиэтилбромид(30,4 г, 0,1 моля) прибавляют к ацетогидроксамовой кислоте (8,5 г,0,13 моля) в 110 мл этанольного раствора гидроксида натрия (4,0 г, 0,1 моля), как описано выше в примере 1, и смесь кипятят с обратным холодильником в течение 6 часов, охлаждают до комнатной температуры,фильтруют, этанол выпаривают и остаток растворяют в ацетоне (100 мл), раствор фильтруют и концентрируют с получением 19,2 г (68 ) 2-(2,4,5-трихлорфенокси)этилацетогидроксамата , т. пл. 160-162 С. Ацетогидроксаматгидролизуют до 2-(2,4,5-трихлорфенокси)этоксиамина гидрохлористого , как описано для соответствующего пропилацетогидроксамата . Этоксамин гидрохлорид подвергают реакции с изопропилдицианодиамидом, как описано ранее в примере 1, с получением -2-(2,4,5-трихлорфенокси)этокси-(1-метилэтил)имидокарбонимидного диамида гидрохлорида . Пример 6.-(2,4,5-трихлорбензокси)(1-метилэтил)имидокарбонимидный диамид гидрохлорид . 2,4,5-Трихлорбензилбромид(16,1 г, 0,1 моля) прибавляют к ацетогидроксамовой кислоте (8,5 г,0,13 моля) в 110 мл этанольного раствора гидроксида натрия (4,0 г, 0,1 моля), как описано ранее в примере 1, и смесь кипятят с обратным холодильником в течение 6 часов, охлаждают до комнатной температуры и фильтруют. Этанол выпаривают, а остаток растворяют в ацетоне (100 мл), раствор фильтруют и концентрируют, получая 2-(2,4,5-трихлорбензи) ацетогидроксамат . Ацетогидроксаматгидролизуют до 2,4,5-трихлорбензоксиамина гидрохлорида , как описано для соответствующего пропилацетогидроксамата . Бензоксиамин гидрохлоридвводят в реакцию с изопропил-дициано-диамидом, как описано ранее в примере 1, с получением -(2,4,5-трихлорбензокси)(1 метилэтил)имидодикарбонимидного диамида гидрохлорида . Пример 7.. п-Хлорфенилизотиоцианат(50,7 г) прибавляют при перемешивании к суспензии цианамида натрия(19,2 г) в этаноле (30 мл), который медленно растворяется и осаждает натриевую соль -цианопхлорфенилтиомочевины (ХХХа), которую отфильтровывают, промывают этанолом и сушат, получая 36,2 г, которые суспендируют в 200 мл этанола и объединяют с 37,6 г метилиодида при быстром перемешивании при комнатной температуре. При выделении тепла продукт отделяется. Суспензию охлаждают в ледяной бане, твердые частицы отфильтровывают, промывают водой и сушат, получая -цианоп-хлорфенилметилизотиомочевину-метилизотиомочевину , приготовленную, как указано выше, прибавляют к этанольному раствору метиламина (79,4 мл, содержащие 4,2 г метиламина) и смесь нагревают 4 часа в толстостенной склянке при 50 С. Получающийся в результате прозрачный раствор постепенно разбавляют водой (75 см 3) и продукт выкристаллизовывается, его отфильтровывают и получают желаемый дициандиамид . В соответствии с вышеприведенной методикой, но там, где вместо метиламина используют соответствующий фенил-, этил-, изопропил-, пропил- и бензиламин, получают соответственно дицианфенил-, этил, изопропил-, пропил- и бензилдиамид. Дицианодиамидзатем подвергают реакции с -3-(2,4,5-трихлорфенокси) пропоксиамином гидрохлоридом , как описано ранее в примере 1, получая титульное соединение. Характеристики других полученных соединений представлены в таблице 8. Пример 8. Фармацевтическая композиция. Таблетки -3-(1,4,5-трихлорфенокси)пропокси(1,3-метилэтил)имидодикарбонимидного диамида гидрохлорида гидрата (табл. 7). Одна таблетка содержит 25 мг - 500 мг активного ингредиента, в зависимости от того, на какой конкретно организм производится воздействие, из-за различной чувствительности болезнетворного микроба. Рецептура разработана для производства 100000 таблеток (15,8-94,8 кг). Таблетки будут покрыты гидроксипропилметилцеллюлозой, красящим веществом, диоксидом титана, полиэтиленгликолем 6000 и карнаубским воском. Вес покрытия составит приблизительно 2-5 от веса таблетки. Таблица 1 Активность РС 7776 против инфекций аМалярийный штамм СМИТ Первичное лечение Излечено 0/4 3/8 6/8 одно животное умерло рано 3/3 Активность прогуанила против инфекций аМалярийный штамм Смит Первичное лечение Излечено 0/2 0/2 0/2 Активность циклогуанила против инфекций аМалярийный штамм Смит Первичное лечение Излечено 0/2 0/2 0/2 Таблица 2 Активность РС 7776, триазина 99210 и прогуанила против инфекций . . Сравнение инъецированных и оральных доз Испытуемое лекарство 50 излечение, инъекцияЕД-50, мг/кг 50 излечение, орально РО ЕД 50, мг/кг 7776 567 (7/10 излечений при 640). Нетоксич 498 но Триазин 99210 245 Отсутствие излечения при 640 Прогуанил Отсутствие излечения Отсутствие излечения. Токсичен при 160 12 3847 1 Таблица 3 Сравнительная эффективность подкожных и оральных доз РС 7776, вводимых мышам, зараженным инфекцией. Более высокийвыживания и излечения Подкожная доза) обозначает активность с выживанием, более чем в 2 раза превышающим выживаемость контрольной группы, или излечениями, основанными на 30-дневном выживании животных. Таблица 4 Потенциирование 7776 сульфонамидами в малярийных паразитах, ингибированных. Фактор потенциирования Паразит Африканский) Фактор потенциирования является отношением 50-ной величины ингибирования (-50) испытуемого лекарства без сульфонамида, деленной на -50, против того же паразита при использовании эквивалентного стандартного количества сульфонамида. Число обработанных особей 10/10 10/10 10/10 10/10 Число инфицированных особей 0/10 0/10 0/10 10/10 3847 1 Таблица 6 Активность РС 7776 и других лекарств против изолятов Микобактериум .,.и . . Концентрации , мкг/мл, для ингибирования ростаИзолят Активный ингредиент Микрокристаллическая целлюлоза Повидон К 29-32 Крахмальный глюконат натрия Стеарат магния Полный вес 100000 таблеток изопропил изопропил изопропил изопропил изопропил изопропил изопропил изопропил изопропил изопропил изопропил изопропил изопропил изопропил изопропил изопропил Элементный анализ Вычислено Найдено С изопропил изопропил изопропил изопропил изопропил Продолжение табл. 8 Элементный анализ Т. пл. С Вычислено Найдено Соль С Государственный патентный комитет Республики Беларусь. 220072, г. Минск, проспект Ф. Скорины, 66.
МПК / Метки
МПК: C07C 321/28, C07C 279/26, A61P 31/08, A61P 33/06, A61K 31/155, A61P 31/04
Метки: антимикробной, снижения, инфекций, диамиды,обладающие, профилактическая, уровня, композиция, n,n'-замещенные, антипаразитической, активностью, имидодикарбонимидные
Код ссылки
<a href="https://by.patents.su/15-3847-nn-zameshhennye-imidodikarbonimidnye-diamidyobladayushhie-antiparaziticheskojj-i-antimikrobnojj-aktivnostyu-profilakticheskaya-kompoziciya-kompoziciya-dlya-snizheniya-urovnya-infek.html" rel="bookmark" title="База патентов Беларуси">N,N’-Замещенные имидодикарбонимидные диамиды,обладающие антипаразитической и антимикробной активностью, профилактическая композиция, композиция для снижения уровня инфекций</a>
Замещенные ациламинобензамиды, обладающие фунгицидной активностью, и способ их получения

Номер патента: 1358
Опубликовано: 16.09.1996
Авторы: Патрик Джелф Кроули, Розамунд Элисон Спенс, Эласдейр Томас Глен
МПК: A01N 37/20, C07C 237/42
Метки: обладающие, получения, способ, фунгицидной, ациламинобензамиды, активностью, замещенные
Текст:
...и сырой 2-хлор-4-нитробензоилхлорид по каплям прибавляют к 40 водному диметиламину(70 мл) при 0-5 С. После перемешивания в течение 0,5 час отфнльтровывают желтый кристаллический осадок, промывают водой, сушат и получают 2-хлор-4-нитро Ы, Ы-диметилбензамид в виде бледно-желтых кристаллов (24,97 г), т. пл. 116-11 ТС.порошкообразное железо (предварительно восстановленное водородом, 10,0 г) суспендируют в этаноле (80 мл) и воде (10 мл) и при...
Производные пропеновой кислоты, обладающие фунгицидной активностью

Номер патента: 1297
Опубликовано: 16.09.1996
Авторы: Поль Джон де Фрейн, Энн Мартин
МПК: C07C 251/40
Метки: производные, фунгицидной, пропеновой, кислоты, обладающие, активностью
Текст:
...вгашйпйв на зерновых. Сегсозрога агасшспсыа и Сегсозрогйайиш регвопата на земляном орехе и другие виды Сегдо рога на других культурах, например сахарной свекле, бананах, соевых бобах и рисе. Вотгупс сшегеа (серая плесень) на томатах,клубнике, овощах, винограде и других культурах, Апегпагйа 5 рр., на овощах (например,огурцах), масличном рапсе, яблоке, томатах и других культурах. Уептвгйа йпаедианв (парша) на яблоне. Ршвшората у 111 со 1 а на...
Производные акриловой кислоты и их стереоизомеры, обладающие фунгицидной активностью
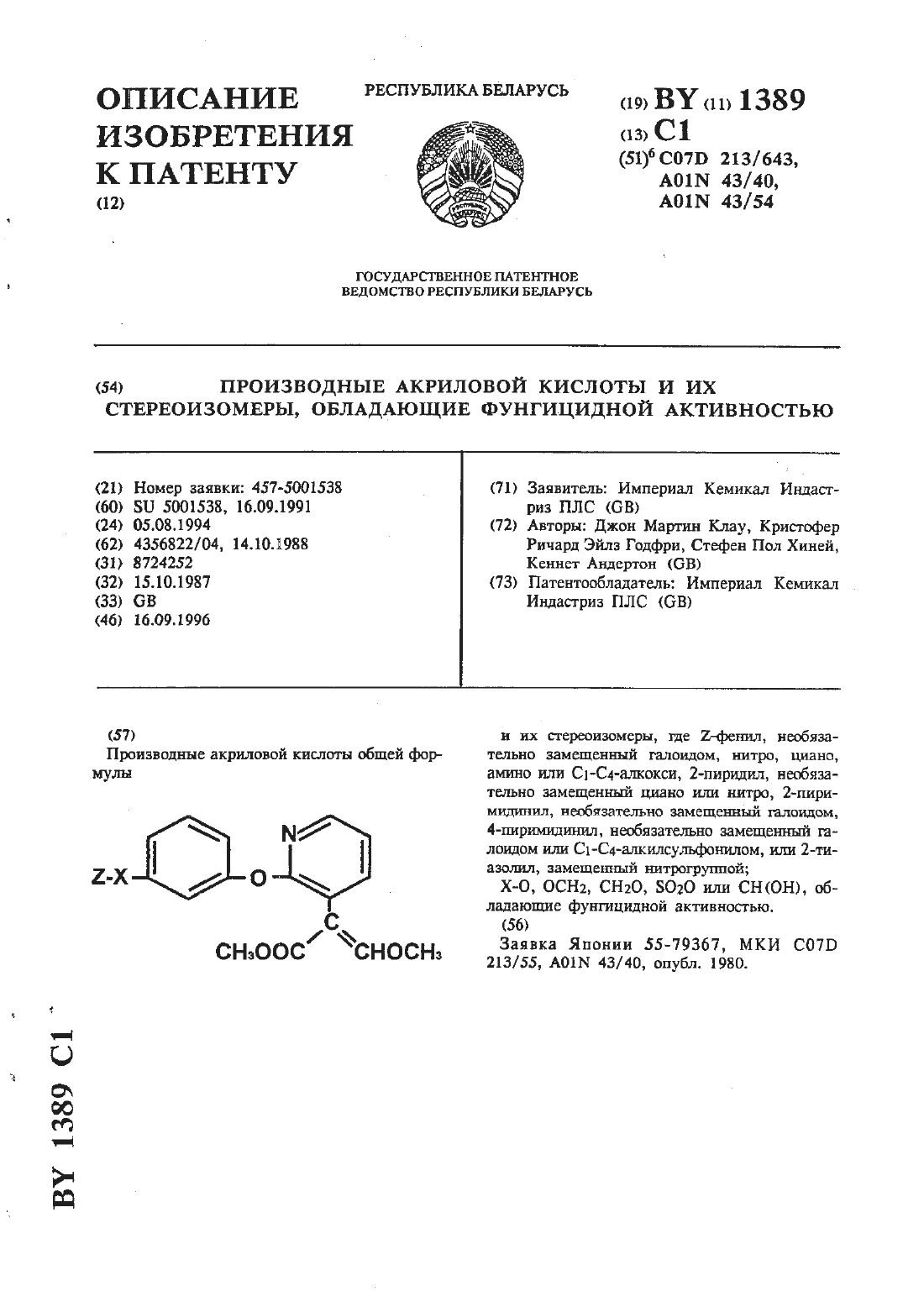
Номер патента: 1389
Опубликовано: 16.09.1996
Авторы: Джон Мартин Клау, Кеннет Андертон, Кристофер Ричард Эйлз Годфри, Стефен Пол Хиней
МПК: C07D 213/643, A01N 43/54, A01N 43/40...
Метки: акриловой, кислоты, стереоизомеры, производные, фунгицидной, обладающие, активностью
Текст:
...минус 7 ОС и обрабатывают, как указано выше во второй загрузке диизобутилалюминнй тилридом (27,6 мл). Через 1 ч по данным газового хроматографического анализа при комнатной температуре отмечают отсутствие исходного материала. Разбавленную хлористоводородную кислоту 60) добавляют чрезвычайно осторожно по причине экзотермической реакции. Полученный раствор перемешивают еще 30 МИН, ЗВТСМ РЗЗДСЛШОТ на СЛОИ ПОСРЕДСТВОМ эфира. Водный слой...
3-циан-5,4′-бипиридин-1′-оксиды и их физиологически совместимые соли присоединения кислот, обладающие кардиотонической и сосудорасширяющей активностью

Номер патента: 1026
Опубликовано: 15.12.1995
Авторы: Готтфрид Фауст, Петер Мушик, Ерхард Клаушенц, Бригитте Шлегель, Ханс-Йоахим Енш, Фолькер Хаген, Ангела Хаген, Сабине Хеер
МПК: C07D 213/89, A61K 31/44
Метки: соли, обладающие, активностью, совместимые, присоединения, кардиотонической, кислот, физиологически, 3-циан-5,4'-бипиридин-1'-оксиды, сосудорасширяющей
Текст:
...из 32.3 г 2-клор-3-циан-5-(пиридил-4-пиридина и 245 мл ледянойуксусной кислоты нагревают на водяной бане при перемешивании при 70 С и осторожно прикалывают 42 мл 40-ной надуксусной кислоты. Образуется светло-желтый прозрачный раствор. После прикапывания температуру водяной бани повышают до 80 С при непрерывном перемешивании. Затем за интервал 1 ч прикалывают еще раз 25 мл 4 О-ной надуксусной кислоты порциями по 5 мл (5 х 5 мл) в смесь....
Аналоги тримера 2′, 5′-олигоадениловой кислоты, обладающие активностью против вирусов, поражающих картофель

Номер патента: 1565
Опубликовано: 30.03.1997
Авторы: Квасюк Евгений Иванович, Зинченко Анатолий Иванович, Барай Владимир Николаевич, Коновалова Галина Ивановна, Сентюрёва Светлана Леонидовна, Кулак Тамара Ивановна, Ященко Нина Петровна, Михайлопуло Игорь Александрович, Ткаченко Ольга Васильевна
МПК: C07H 19/20, A01N 57/16
Метки: кислоты, картофель, обладающие, вирусов, тримера, против, аналоги, 5'-олигоадениловой, активностью, поражающих
Текст:
...75 мл хлороформа и обрабатывают ее 0,05 М раствором ТЕАБ (2 х 20 мл). Органический слой отделяют, сушат безводным сульфатом натрия и упаривают досуха. Остаток хроматографируют на силикагеле (75 см 3). Продукты реакции элюируют смесью растворителей хлороформ-метанол в градиенте концентрации метанола от 0,1 до 2 объемных . Фракции, содержащие блокированный тример , объединяют, упаривают до объема 3 мл и высаждают в 100 мл гексана. Получают в...
Предыдущий патент: Способ получения армированной полиамидной композиции
Следующий патент: Способ получения порошковых покрытий
Случайный патент: Способ лечения отгематомы