Способ определения преимущественных направлений мутагенеза в гене ДНК- или РНК-вируса, поражающего человека
Номер патента: 17123
Опубликовано: 30.06.2013
Авторы: Хрусталев Владислав Викторович, Ермолович Марина Анатольевна, Самойлович Елена Олеговна, Семейко Галина Валерьевна






Текст
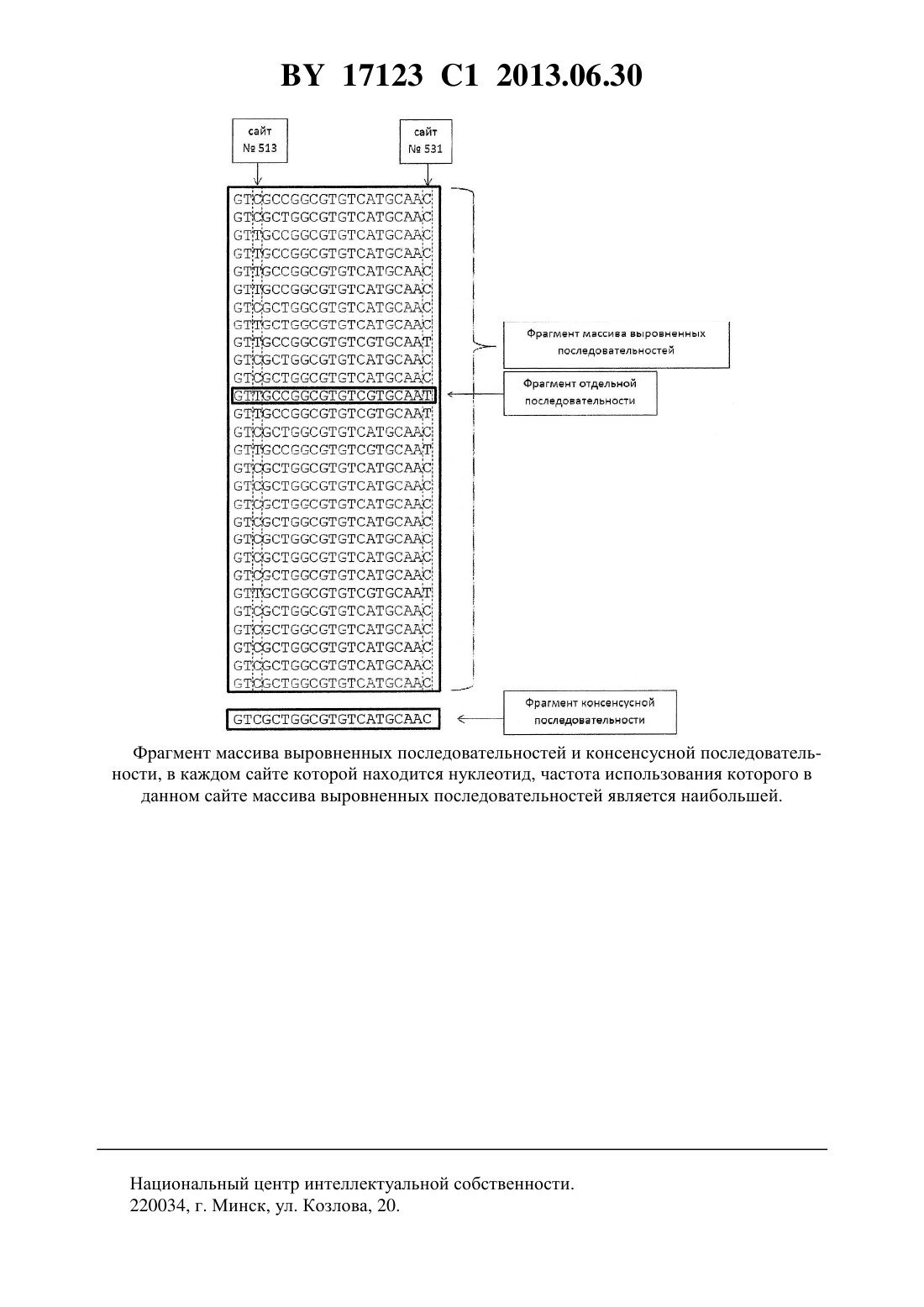
(51) МПК НАЦИОНАЛЬНЫЙ ЦЕНТР ИНТЕЛЛЕКТУАЛЬНОЙ СОБСТВЕННОСТИ СПОСОБ ОПРЕДЕЛЕНИЯ ПРЕИМУЩЕСТВЕННЫХ НАПРАВЛЕНИЙ МУТАГЕНЕЗА В ГЕНЕ ДНК- ИЛИ РНК-ВИРУСА,ПОРАЖАЮЩЕГО ЧЕЛОВЕКА(71) Заявитель Учреждение образования Белорусский государственный медицинский университет(72) Авторы Хрусталев Владислав Викторович Самойлович Елена Олеговна Ермолович Марина Анатольевна Семейко Галина Валерьевна(73) Патентообладатель Учреждение образования Белорусский государственный медицинский университет(56).,1996. - . 2.. 7. - . 753-759.2172015 1, 2001. БАРКОВСКИЙ Е.В. и др. Актуальные вопросы молекулярной эволюции и биохимии. Материалы Республиканской научной конференции, посвященной 75-летию со дня основания кафедры общей химии БГМУ. - Минск, 2006. С. 27-30. ХРУСТАЛЕВ В.В. и др. Медицинский журнал. - 2006. -2. - С. 101-105. ХРУСТАЛЕВ В.В. и др. Медицинский журнал. - 2006. -4. - С. 99-103. ХРУСТАЛЕВ В.В. и др. Медицинский журнал. - 2007. -1. - С. 92-96. БУТВИЛОВСКИЙ А.В. и др. Здравоохранение. - 2005. -6. - С. 37-39 БУТВИЛОВСКИЙ А.В. и др. Здравоохранение. - 2005. -6. - С. 40-43.(57) Способ определения преимущественных направлений мутагенеза в гене ДНК- или РНК-вируса, поражающего человека, заключающийся в том, что выделяют исследуемый вирусный ген из образцов клинического материала от разных больных, проводят амплификацию нуклеотидных последовательностей, кодирующих исследуемый вирусный ген,секвенируют полученные нуклеотидные последовательности, выравнивают их, рассчитывают процентное содержание нуклеотидов , ,идля ДНК-вируса или , ,идля РНК-вируса в первом, втором и третьем положениях кодонов полученных нуклеотидных последовательностей, а также частоту встречаемости в сайтахвыровненных нуклеотидных последовательностей каждого из указанных нуклеотидов по формуле 100 где- количество выровненных нуклеотидных последовательностей, содержащих в сайте данный нуклеотид- количество выровненных нуклеотидных последовательностей,на основании полученных значенийсоздают консенсусную нуклеотидную последовательность исследуемого вирусного гена, по отношению к консенсусной последовательности рассчитывают количество сайтов, содержащих нуклеотидную замену, во всех выровненных 17123 1 2013.06.30 последовательностях в случае ДНК-вируса или количество выровненных последовательностей, содержащих нуклеотидную замену, в случае РНК-вируса и определяют, что мутагенез исследуемого вирусного гена предпочтительно направлен на частую нуклеотидную заменунаилина , если установлено, что количество заменнаинаилинаинапревышает количество заменнаина , а содержаниев третьих положениях кодонов выровненных нуклеотидных последовательностей более 25 илинаилина , если установлено, что количество заменнаинаилинаинапревышает количество заменнаина , а содержаниеилив третьих положениях кодонов выровненных нуклеотидных последовательностей более 25 илина , если установлено, что количество заменнаинапревышает количество заменнаинаилинаина , а содержаниев третьих положениях кодонов выровненных нуклеотидных последовательностей более 25 илина , если установлено, что количество заменнаинапревышает количество заменнаинаилинаина , а содержаниев третьих положениях кодонов выровненных нуклеотидных последовательностей более 25 илина , если количество этих замен или заменнапревышает количество заменнаинаилинаинаилинаилина , если количество этих заменнаинаили заменнаинапревышает количество заменнаина . Изобретение относится к медицине, а именно к медицинской вирусологии и молекулярной биологии, может быть использовано для определения механизма мутагенеза в генах вирусов, поражающих человека, с целью последующего использования полученных данных для выделения наименее мутабельных иммуногенных фрагментов белков или предотвращения гипермутабельности вирусов. Известен способ определения точечных нуклеотидных замен в ДНК микобактерий 1. С помощью данного способа можно определить наличие определенного числа нуклеотидных мутаций во фрагменте гена(ДНК-зависимой РНК-полимеразы) микобактерии туберкулеза, предположить их направление и механизм. Способ основан на амплификации фрагмента генаизучаемого штамма микобактерии туберкулеза с помощью полимеразной цепной реакции (ПЦР) с последующей гибридизацией амплифицированной ДНК на биочипе, содержащем определенный набор олигонуклеотидов, с которыми может происходить комплементарное связывание образца. Факт комплементарного связывания амплифицированного фрагмента изучаемого гена с каким-либо олигонуклеотидом распознается по изменению флуоресценции. Основным недостатком данного способа является то, что набор нуклеотидных мутаций, которые он позволяет выявить, ограничен. Кроме того, он может быть применен исключительно к фрагменту генамикобактерии туберкулеза. Судить о направлении и механизме мутации можно путем сопоставления собственных данных с последовательностью гена, принятой за исходную, что не всегда соответствует действительности. Наиболее близким способом, принятым за прототип, является способ выявления мутаций при помощи гибридизации на олигонуклеотидных микроматрицах 2. Сущность способа заключается в том, что производят выделение нуклеотидных последовательностей исследуемого вирусного гена из образцов клинического материала, который подвергается амплификации при помощи ПЦР, а затем происходит фрагментация (случайным образом ДНК нарезается на участки одинаковой длины). После этого полученный набор участков ДНК связывается с меченными флуоресцентными зондами олигонуклеотидными последовательностями, закрепленными на специальной микроматрице. На матрице должны присутствовать все возможные комбинации из восьми нуклеотидов (всего 65 536 олигонуклеотидов). Комплементарное связывание одного из участков изучаемого гена с каким-либо олигонуклеотидом распознается по изменению флюоресценции. Получение 2 17123 1 2013.06.30 данных о мутагенезе исследуемого вируса производят путем сравнения с последовательностью гена, принятой за исходную. Известный способ имеет ряд недостатков. В результате анализа участка гена , кодирующего протеазу (297 нуклеотидов длиной), 114 штаммов ВИЧ 1 с помощью гибридизации на олигонуклеотидной микроматрице было выявлено 98,26 мутаций, по сравнению с результатами прямого секвенирования. Чем длиннее анализируемый участок гена, тем ниже процент выявления мутаций в нем данным методом (для последовательности длиной 5 400 нуклеотидов - только 74 ). На качестве анализа также отрицательно сказывается наличие прямых и инвертированных повторяющихся последовательностей в изучаемом фрагменте ДНК или РНК (образуются вторичные структуры, препятствующие связыванию с олигонуклеотидами). Всех этих недостатков можно избежать при использовании прямого секвенирования для последующего анализа механизмов и направлений мутаций в вирусных генах. Кроме того, для анализа путем гибридизации на олигонуклеотидной микроматрице какой-либо вариант вирусного гена необходимо принимать за исходный, чтобы по отношению к нему определять направление мутации. В предложенном нами способе этот недостаток устраняется путем создания консенсусной последовательности по результатам секвенирования определенного количества (более двух) последовательностей. Задачей заявляемого способа является увеличение точности выявления мутации в вирусных генах, повышение эффективности анализа их количества и направления, а следовательно, определение наиболее вероятного биохимического механизма, вызывающего их. Поставленная задача достигается с помощью предлагаемого способа определения преимущественных направлений мутагенеза в гене ДНК- или РНК-вируса, поражающего человека, заключающегося в том, что выделяют исследуемый вирусный ген из образцов клинического материала от разных больных, проводят амплификацию нуклеотидных последовательностей, кодирующих исследуемый вирусный ген, секвенируют полученные нуклеотидные последовательности, выравнивают их, рассчитывают процентное содержание нуклеогидов , ,идля ДНК-вируса или , ,идля РНК-вируса в первом,втором и третьем положениях кодонов полученных нуклеотидных последовательностей, а также частоту встречаемости в сайтахвыровненных нуклеотидных последовательностей каждого из указанных нуклеотидов по формуле 100, где- количество выровненных нуклеотидных последовательностей, содержащих в сайте данный нуклеотид- количество выровненных нуклеотидных последовательностей,на основании полученных значенийсоздают консенсусную нуклеотидную последовательность исследуемого вирусного гена, по отношению к консенсусной последовательности рассчитывают количество сайтов, содержащих нуклеотидную замену, во всех выровненных последовательностях в случае ДНК-вируса или количество выровненных последовательностей, содержащих нуклеотидную замену, в случае РНК-вируса и определяют, что мутагенез исследуемого вирусного гена предпочтительно направлен на частую нуклеотидную заменунаилина , если установлено, что количество заменнаинаилинаинапревышает количество заменнаина , а содержаниев третьих положениях кодонов выровненных нуклеотидных последовательностей более 25 илинаилина , если установлено, что количество заменнаинаилинаинапревышает количество заменнаина , а содержаниеилив третьих положениях кодонов выровненных нуклеотидных последовательностей более 25 илина , если установлено, что количество заменнаинапревышает количество заменнаинаилинаина , а содержаниев третьих положениях кодонов выровненных нуклеотидных последовательностей более 253 17123 1 2013.06.30 илина , если установлено, что количество заменнаинапревышает количество заменнаинаилинаина , а содержаниев третьих положениях кодонов выровненных нуклеотидных последовательностей более 25 илина , если количество этих замен или заменнапревышает количество заменнаинаилинаинаилинаилина , если количество этих заменнаинаили заменнаинапревышает количество заменнаина . Пример 1. Определение основных механизмов мутагенеза вируса краснухи. Для выделения вируса краснухи использовали носоглоточные мазки и пробы мочи от больных краснухой, чей диагноз подтвержден лабораторно путем выявления в сыворотке крови специфических -антител к вирусу краснухи в иммуноферментной тест-системе производства - (Германия). Носоглоточные мазки и пробы мочи были собраны в течение 2004-2006 гг. у больных, проживающих в Минске, Минской области, Витебске и Могилеве. Изоляцию вируса проводили в перевиваемой культуре клеток . Вирусную РНК экстрагировали из 140 мкл вируссодержащей культуральной жидкости с использованием набора(, Нидерланды) согласно инструкции производителя. Обратную транскрипцию проводили в 20 мкл реакционной смеси, содержащей 1 мкл обратной транскриптазы(200 ед.), 0,5 мкл ингибитора рибонуклеаз(20 ед.), 2 мкл 0,1, 4 мкл 5 буфера, 1 мкл 10 мМ смеси дНТФ (, Бельгия), 5 мкл 0,03 мкг/мкл универсальных гексамеров(, Бельгия), 1,5 мкл воды и 5 мкл выделенной РНК. Начальную денатурацию выполняли при 65 С в течение 5 мин при отсутствии ферментов, реакцию обратной транскрипции - 80 мин при 55 С и 15 мин при 70 С. С целью амплификации -гена использовали праймеры производства(Бельгия). Концентрация всех праймеров составляла 40 мкМ. ПЦР проводили в 25 мкл реакционной смеси, содержащей 0,1 мкл ДНК полимеразы(5 ед.), 0,5 мкл 10 мМ смеси дНТФ, 2,5 мкл 10 буфера,1 мкл 50 мМ 2 (, Бельгия), по 0,25 мкл каждого из праймеров, 15,4 мкл воды и 5 мкл ДНК, комплементарной вирусной РНК (кДНК). Реакцию проводили в следующих условиях начальная денатурация кДНК - 94 С (3 мин), 40 циклов амплификации, включающих денатурацию - 94 С (30 с), отжиг праймеров - 61 С (30 с), элонгацию - 72 С(45 с). В последнем цикле амплификации элонгацию проводили в течение 5 мин. Для повышения чувствительности реакции 1 мкл смеси, после первого раунда ПЦР, использовали для постановки гнездовой ПЦР. Амплификацию проводили аналогичным образом. Синтез ПЦР-продуктов анализировали методом электрофореза в 1,5 агарозном геле в трис-ацетатном буфере с добавлением бромистого этидия (, Бельгия). Перед проведением секвенирования полученные фрагменты ДНК очищали из смеси с использованием набора(, Германия) или из геля с помощью набора(, Нидерланды). Секвенирование нуклеотидных последовательностей проводили с использованием набора.3.1( , Нидерланды) на капиллярном секвенаторе 3100 ( , Нидерланды). Были просеквенированы 28 нуклеотидных последовательностей. Для 13 штаммов вируса краснухи определены нуклеотидные последовательности полноразмерного 1-гена(длиной 1443 нуклеотида), кодирующего поверхностный гликопротеин, еще для 15 штаммов просеквенирован фрагмент 1-гена (длиной 739 нуклеотидов). Нуклеотидные последовательности вируса краснухи обогащены цитозином в третьих положениях кодонов (содержание цитозина в третьих положениях кодонов варьируется от 48,6 до 51,6 ). Полученные нуклеотидные последовательности обеднены аденином и урацилом в третьих положениях кодонов (частоты использования данных нуклеотидов варьировались от 4 17123 1 2013.06.30 6,5 до 7,3 и от 12,6 до 16,4 соответственно). Уровень гуанина в третьих положениях кодонов был ниже уровня цитозина (от 28,5 до 32,0 ). Полученные данные позволяют сделать вывод о том, что в геноме вируса краснухи, представленном одноцепочечнойРНК,существует -давление преобладают замены, повышающие содержание цитозина 1, 2. Пример расчета процентного содержания цитозина в сайте 513 (фигура) выровненных последовательностей по формуле 2100 18 10064,29 28 В каждом сайте выровненных последовательностей 5, 6 проверялось следующее условие нуклеотид, находящийся в данном сайте консенсусной последовательности должен быть . Далее производился подсчет последовательностей, в данном сайте которых находится . В конечном итоге было вычислено количество заменнадля всех сайтов,в консенсусной последовательности которых находится . Например, в сайте 513 консенсусной последовательности (фигура) находится , количество последовательностей,содержащих в данном сайте , составляет 10. В сайте 531 консенсусной последовательности (фигура) находится , количество последовательностей, содержащих в данном сайте , составляет 5. Количество заменнадля этих двух сайтов равно 15. Количество заменнадля всех сайтов равно 224. Количество заменнаравно 167. Поскольку уровень 3 составляет примерно 50 , логично предположить, что в действительности большая часть таких замен имела направлениена , но частоты какой-то части из них в общей популяции достигли уровня выше 50 , из-за чего при определении направления замены от консенсусной последовательности им было приписано противоположное направление. На втором месте по частоте - заменына(64 замены) ина(47 замен). Поскольку уровень использования гуанина в третьих положениях кодонов превышает 25 , следует признать, что вторым наиболее вероятным механизмом мутаций в генах вируса краснухи являются транзицийна . Наиболее вероятным активным механизмом и тех и других транзиции является дезаминирование аденина с образованием инозина, который комплементарен цитозину, а не урацилу. Заменынапроисходят за счет дезаминирования аденина на РНКцепи вируса. Количество трансверсий было значительно ниже, чем количество транзиций. Тем не менее, судя по соотношению количества трансверсий разных типов (/0,741),гуанин несколько чаще окисляется на РНКцепи вируса. Мутагенез вируса краснухи можно охарактеризовать следующим образом. Наиболее частым механизмом мутаций является дезаминирование аденина, которое вызывается клеточными РНК-редактирующими ферментами из семейства . Дезаминирование аденина происходит чаще на РНКцепи вируса, в отличие от окисления гуанина, которое чаще происходит на РНКцепи. Этими двумя обстоятельствами можно объяснить тот факт, что частота использования цитозина в геномной РНКцепи выше частоты использования гуанина. Пример 2. Определение основных механизмов мутагенеза парвовируса 19. Геном парвовируса 19 представлен одноцепочечной ДНК. Материалом для получения ДНК парвовируса 19 служили сыворотки крови больных с парвовирусной инфекцией,проживающих в г. Минске, Минской, Брестской, Витебской, Могилевской, Гродненской и Гомельской областях, собранные в течение 2006-2007 гг. Вирусную ДНК выделяли из 200 мкл сыворотки крови с помощью набора(Нидерланды) согласно инструкции производителя. Амплификацию фрагмента ДНК, кодирующего 1/1 область генома (длиной 994 нуклеотида), проводили методом гнездовой ПЦР. Для первого раунда использовали праймеры 1855 и 19-1, для второго - 1863 и 192 концентрацией 20 мкМ. Объем реакционной смеси составлял 25 мкл, содержащей 0,1 мкл ДНК полимеразы(5 ед.), 0,5 мкл 10 мМ смеси дНТФ, 2,5 мкл 10 5 17123 1 2013.06.30 буфера, 1 мкл 50 мМ 2 (, Бельгия), по 1 мкл каждого из праймеров, 13,9 мкл воды и 5 мкл выделенной ДНК. Реакцию проводили в следующих условиях начальная денатурация - 5 мин при 94 С, затем 40 циклов амплификации 30 с при 94 С, 30 с при 55 С и 1 мин 30 с при 72 С, завершающая элонгация - 5 мин при 72 С. Для второго раунда ПЦР использовали 0,5 мкл продукта, полученного в первом раунде реакции. Амплификацию проводили аналогично, повысив температуру отжига праймеров до 58 С. Синтез ПЦР-продуктов анализировали методом электрофореза в 1,5 агарозном геле в 1 буфере. Перед проведением секвенирования полученные фрагменты ДНК очищали из смеси с использованием набора(, Германия) или из геля с помощью набора(, Нидерланды). Секвенирование нуклеотидных последовательностей проводили с использованием набора.3.1( , Нидерланды) на капиллярном секвенаторе 3100 ( , Нидерланды). Были определены нуклеотидные последовательности 1/1 области генома 92 штаммов парвовируса 19, однако для данного исследования использован фрагмент гена (длиной 486 нуклеотидов), кодирующего структурный белок 1. Ниже приведены результаты расчета частот использования нуклеотидов в третьих положениях кодонов- от 15,4 до 16,6- от 11,8 до 13,0- от 42,0 до 40,7- от 30,5 до 30,2 . Общее количество нуклеотидных замен от консенсусной последовательности, установленной на основании расчетов по формуле, составило 31. Определение количества сайтов, содержащих заменуна , проходило при соблюдении следующих условий в данном сайте консенсусной последовательности должен находиться , хотя бы в одной последовательности в данном сайте должен находиться . В изученной выборкеприсутствовал в девяти сайтах, в консенсусной последовательности которых находилсяв сайтах 10, 17, 28, 34, 72, 82, 87, 181, 337. Это говорит о том, что в выборке нуклеотидных последовательностей, кодирующих фрагмент 1 парвовируса 19, было 9 заменна . Заменнабыло 6. Количество замен противоположных направлений было ниже 7 заменнаи 4 заменына . Поскольку частоты использования как тимина, так и аденина в третьих положениях кодонов превышают 25 , во фрагменте гена 1 парвовируса 19 преобладают транзициинаина . Общее количество трансверсий (5 замен) было чрезвычайно низким. Мутагенез парвовируса 19, геном которого представлен одноцепочечной ДНК, можно охарактеризовать следующим образом. Наиболее частый механизм возникновения мутаций - дезаминирование цитозина, происходящее на обеих цепях ДНК, приводящее к транзициямнаина . Дезаминирование цитозина, как правило, вызывается ферментами из суперсемейства . Предложенный способ позволяет повысить точность выявления мутаций в вирусных генах и эффективно производить анализ их количества и направления с целью установления наиболее вероятной биохимической причины мутаций в генах определенного вида вирусов сведения, полученные данным способом, могут быть использованы для борьбы с вирусными заболеваниями с учетом данных о механизмах гипермутабельности вирусов. Источники информации 1.217201501, 1 2001. 2.. - 1996. - .2. - . 7. - . 753-759. 6 Фрагмент массива выровненных последовательностей и консенсусной последовательности, в каждом сайте которой находится нуклеотид, частота использования которого в данном сайте массива выровненных последовательностей является наибольшей. Национальный центр интеллектуальной собственности. 220034, г. Минск, ул. Козлова, 20. 7
МПК / Метки
Метки: определения, преимущественных, или, поражающего, гене, мутагенеза, способ, рнк-вируса, днк, человека, направлений
Код ссылки
<a href="https://by.patents.su/7-17123-sposob-opredeleniya-preimushhestvennyh-napravlenijj-mutageneza-v-gene-dnk-ili-rnk-virusa-porazhayushhego-cheloveka.html" rel="bookmark" title="База патентов Беларуси">Способ определения преимущественных направлений мутагенеза в гене ДНК- или РНК-вируса, поражающего человека</a>
Средство для подавления вируса иммунодефицита человека в культуре клеток

Номер патента: 12529
Опубликовано: 30.10.2009
Авторы: Мельнова Наталья Ивановна, Подольская Ирина Александровна, Рытик Петр Григорьевич, Сорокин Виктор Леонидович, Кучеров Игорь Иванович, Мистрюкова Любовь Олеговна, Полозов Генрих Иванович
МПК: C07C 321/00, A61K 31/095
Метки: средство, подавления, иммунодефицита, культуре, клеток, человека, вируса
Текст:
...и химиотерапевтический индекс(ХТИ) - соотношение МПК/ЕС 50, характеризующее широту спектра его нетоксических эффективных концентраций. Показатель соотношения МПК/50 менее 2,0 указывал на 3 12529 1 2009.10.30 низкую, 2,1-7,9 - на среднюю и более 8,0 - на высокую антивирусную активность препаратов 9. В результате испытаний установлено, что ВИЧ-ингибирующая активность соединения в зависимости от условий проведения экспериментов...
Устройство для определения степени гидратации периферических тканей организма человека и способ ее определения

Номер патента: 14099
Опубликовано: 28.02.2011
Авторы: Шотт Александр Владимирович, Казущик Василий Леонович, Василевич Александр Павлович, Протасевич Алексей Иванович
МПК: A61B 5/00
Метки: способ, устройство, определения, степени, гидратации, человека, тканей, организма, периферических
Текст:
...пациента,сдавленных между эластичной мембраной 14 камеры 6 и прижимным винтом 8 корпуса 1 под постоянным давлением, контролируемым манометром 3. Поскольку камера аппарата,соединительная трубка и манометр представляют собой замкнутую систему, постольку выдавливание жидкости из зажатых тканей сопровождается понижением давления в замкнутой системе. Такое снижение фиксируется с количественной оценкой по манометру во временном интервале до...
Ингибитор вируса герпеса и способ подавления размножения вируса герпеса

Номер патента: 2085
Опубликовано: 30.03.1998
Авторы: Николаева Светлана Николаевна, Хмара Марк Ефимович, Бореко Евгений Иванович
МПК: C12N 7/04, A61K 31/38
Метки: вируса, подавления, ингибитор, способ, размножения, герпеса
Текст:
...тинидазола и ацикловира. При этом используют концентрации препаратов, равные или не достигающие их среднеэффективной концентрации (концентрации, подавляющей размножение вируса на 50) при индивидуальном применении. Полученные результаты сводят в таблицу 2. Таблица 1 Подавление размножения вирусов герпеса простого в культуре клетокпод влиянием тинидазола (Т), ацикловира (АЦГ) и фосфоноуксусной кислоты (ФУК) Препарат Вирусы герпеса и...
Способ определения концентрации в биологической жидкости человека растворимого рецептора интерлейкина-8 второго типа

Номер патента: 15394
Опубликовано: 28.02.2012
Авторы: Котлинский Константин Вячеславович, Войтенок Николай Николаевич, Дорошенко Татьяна Михайловна, Акалович Светлана Тадеушевна, Котлинская Юлия Владимировна
МПК: C07K 16/18, C07K 14/435, G01N 33/577...
Метки: интерлейкина-8, жидкости, концентрации, способ, человека, типа, биологической, второго, рецептора, определения, растворимого
Текст:
...которого идентична аминокислотной последовательности конца рецептора 2 человека от аминокислотного остатка 1 до аминокислотного остатка 20 и дополнительно содержит модифицированный биотином по -группе остаток цистеина на С-конце, и моноклональными мышиными антителами А, специфичными к концевой части рецептора 2, проводят реакцию с тетраметилбензидином, измеряют оптическую плотность продукта реакции, по предварительно построенной...
Способ определения стрессовой реакции у человека

Номер патента: 11408
Опубликовано: 30.12.2008
Авторы: Сидоренко Георгий Иванович, Воробьев Анатолий Павлович, Комисарова Светлана Михайловна, Фролов Александр Владимирович
МПК: A61B 5/02
Метки: определения, реакции, человека, стрессовой, способ
Текст:
...ВСР проводят на аппаратно-программном комплексе Бриз, который позволяет одновременно регистрировать показатели временного, геометрического и спектрального анализа ВСР. При анализе ВРС определяют показатели временных характеристик ЗВЫМ, мс - стандартное отклонение величин интервалов КН за изучаемый период, отражает общую вариабельность ритма гМ 55 В, мс - среднеквадратичное отклонение абсолютных приращений...
Предыдущий патент: Способ и устройство для изготовления стального баллончика с открытым завальцованным концом
Следующий патент: Способ экструзионного гранулирования
Случайный патент: Оборотно-перекидной календарь