9-O-Оксимовые производные эритромицина А и фармацевтическая композиция
Номер патента: 4962
Опубликовано: 30.03.2003
Авторы: БОТТА, Даниела, ПЕЛЛАЧИНИ, Франко, АЛЬБИНИ, Энрико, СКОППАКАССИ, Джованна, РОМАНЬЯНО, Стефано, САНТАНЖЕЛО, Франческо






















Текст
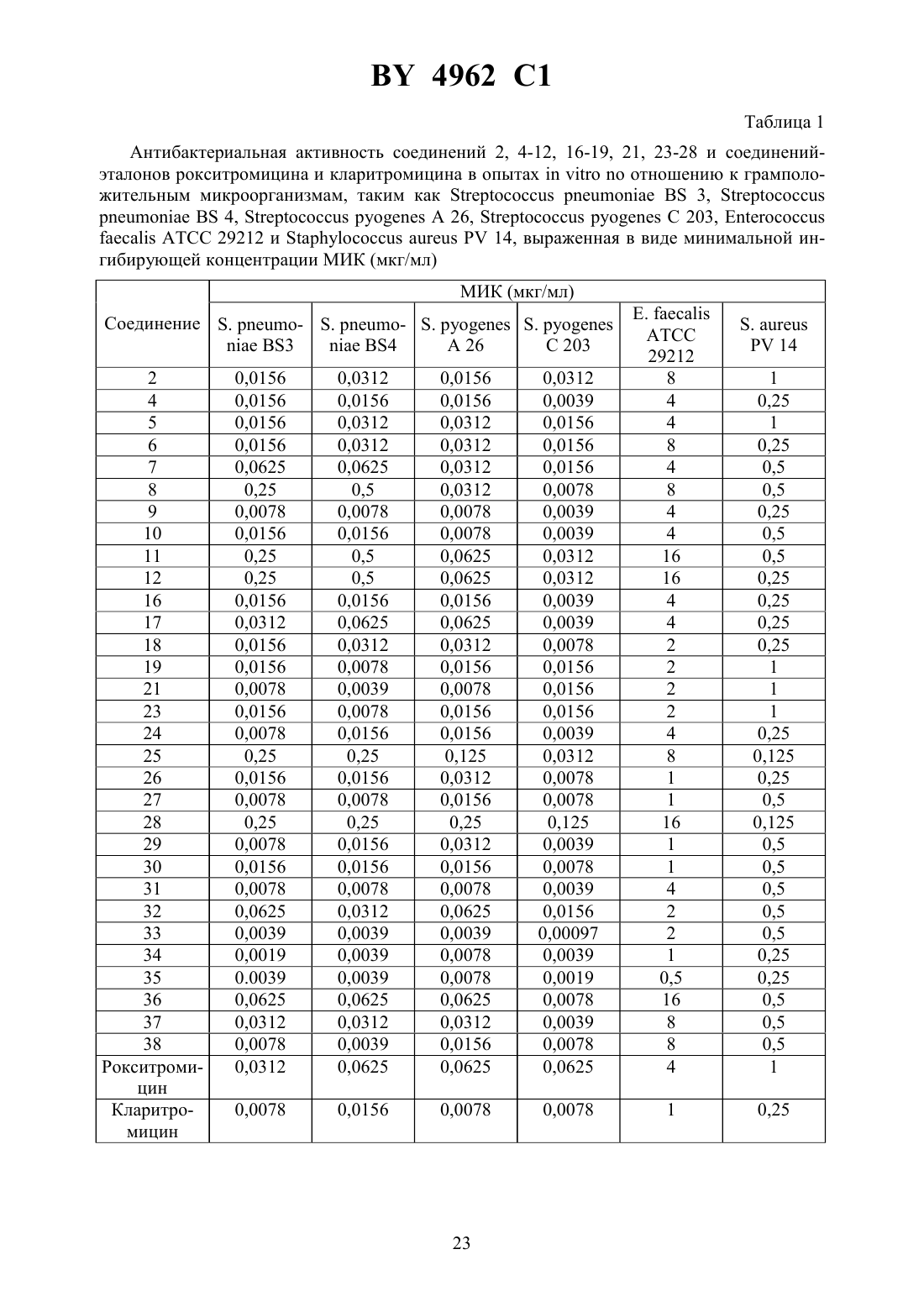
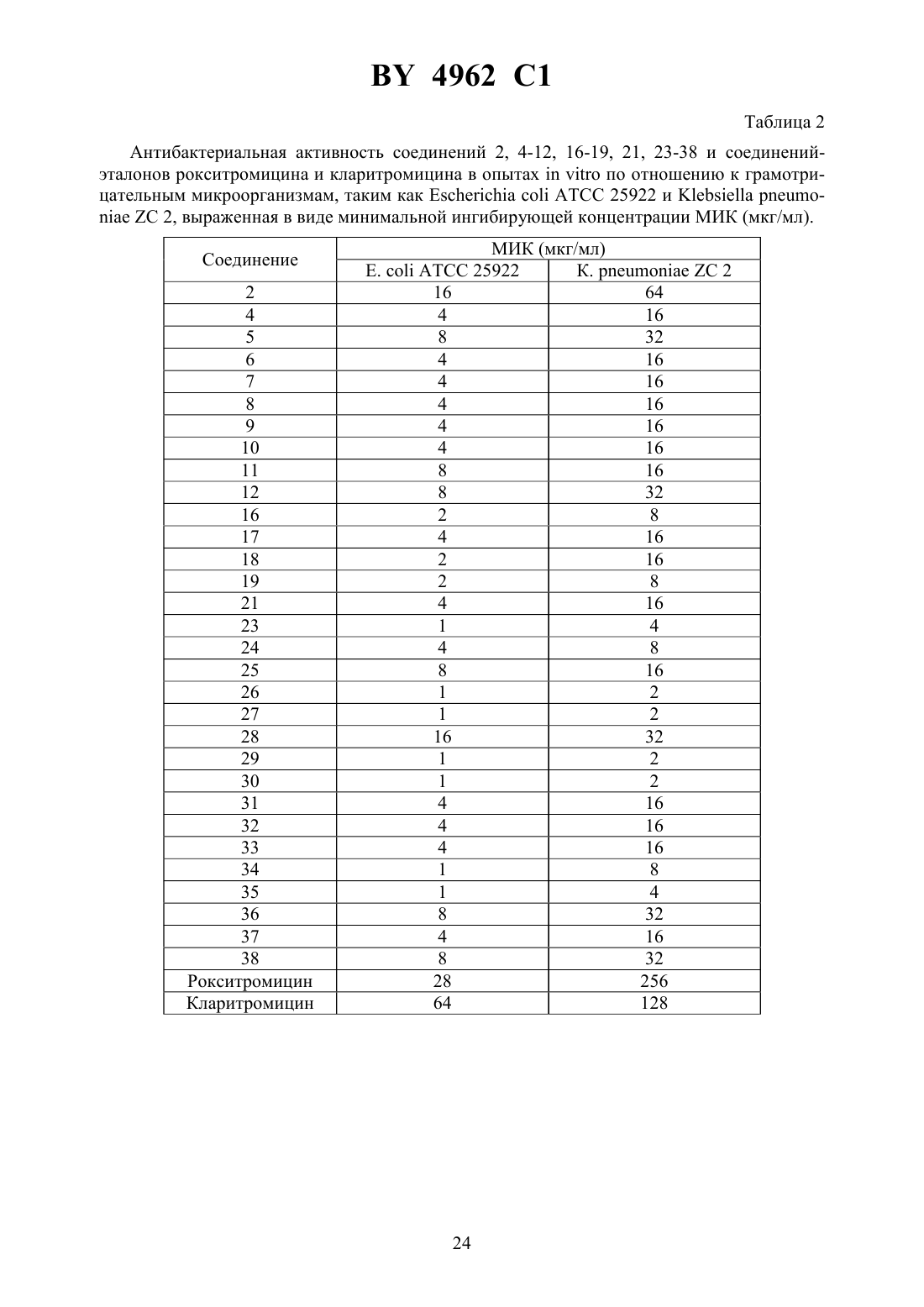
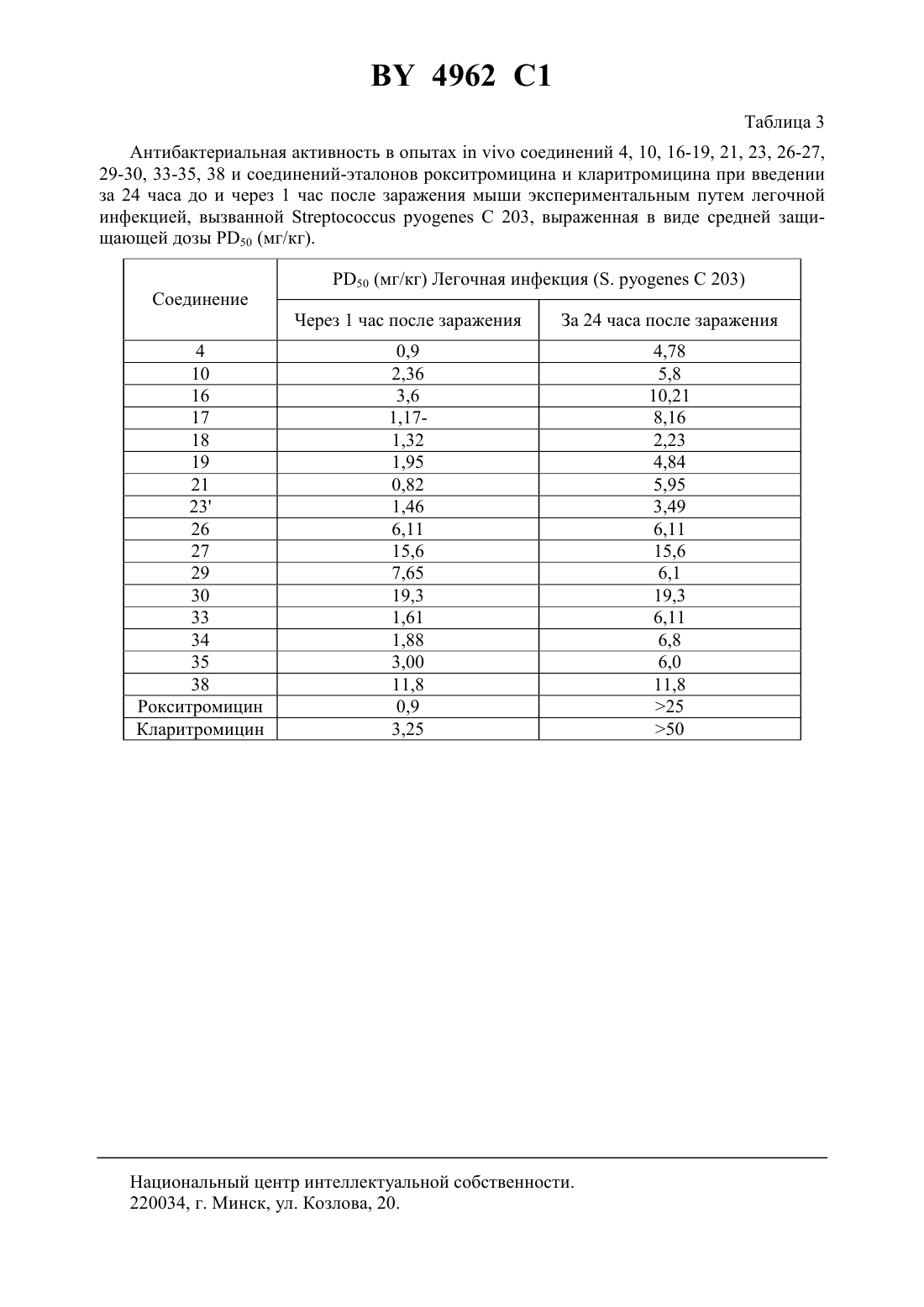
61 31/7048, 61 31/04 НАЦИОНАЛЬНЫЙ ЦЕНТР ИНТЕЛЛЕКТУАЛЬНОЙ СОБСТВЕННОСТИ(71) Заявитель Замбон Груп С.П.А.(73) Патентообладатель Замбон Груп С.П.А. где А обозначает фенил, необязательно замещенный 1-3 одинаковыми или различными группами, выбранными из прямых или разветвленных С 1-С 4-алкокси, С 1-С 2-алкилендиокси, С 1-С 4-алкилсульфонила, фенила, фенокси, гидрокси, нитро, галогена и трифторметила, пиридил или фурил 1 и 2 одинаковые или различные, обозначают водород или прямой или разветвленный С 1-С 4-алкил- 1 или 2- целое число 1 - 8- целое число 2 - 6 М - группа формулы 3 где 3 - водород,или их фармацевтически приемлемые соли. 4962 1 2. Соединение общей формулыпо п. 1, имеющее Е-конфигурацию. 3. Соединение общей формулыпо п. 1, где А - пиридил, фурил или фенил, необязательно замещенный 1 - 3 группами, выбранными из гидрокси, метокси, метилендиокси, нбутокси, фенокси, фенила, метилсульфонила, нитро, галогена и трифторметила 1 и 2 одинаковые или различные, обозначают водород или метил. 4. Соединение общей формулыпо п. 1, где А обозначает фенил, необязательно замещенный группой, выбранной из фенокси, нитро и трифторметила 1 и 2 одинаковые и обозначают водород или метил- 1,- 6- 2. 5. Фармацевтическая композиция, обладающая антибиотической активностью, содержащая терапевтически эффективное количество производного эритромицина А в смеси с фармацевтически приемлемым носителем, отличающаяся тем, что в качестве производного эритромицина А она содержит 9-О-оксимовое производное эритромицина А общей формулыпо п. 1. Настоящее изобретение относится к производным эритромицина А, обладающим антибиотической активностью, пригодным для лечения инфекционных заболеваний, в частности изобретение относится к производным 9-О-(аминоалкил)оксима эритромицина А,обладающим антибиотической активностью по отношению к грамположительным и грамотрицательным микроорганизмам. Эритромицин,изд.,3626 представляет собой хорошо известный, встречающийся в естественных условиях макролид, который обладает антибиотической активностью и который имеет следующую структуру 3 3 Помимо активности по отношению к некоторым микроорганизмам, не относящимся к бактериям, таким как риккетсии и микоплазмы, эритромицин А обладает антибактериальной активностью в основном по отношению к грамположительным микроорганизмам,таким как стрептококки, стафилококки и пневмококки, но он также проявляет эффективность по отношению к некоторым грамотрицательным микроорганизмам, таким как, например,,и. Помимо хорошо известной активности по отношению к вышеуказанным прокариотам по появившимся недавно литературным данным эритромицин А и другие антибиотики из 2 4962 1 класса макролидов обладают активностью по отношению к эукариотичным паразитам и др.,, 28, 307-343 (1994). По отношению к эритромицину А, равно как и по отношению к другим антибактериальным лекарствам, у некоторых штаммов бактерий наблюдали явление устойчивости. В частности, это явление наблюдали при продолжительном применении эритромицина А при лечении инфекций, вызванных стафилококками .иМсК. ,,,, изд., (1987) 851-882. Интерес к антибиотикам из класса макролидов обусловлен возможностью их применения в клинической терапии и ветеринарии при лечении некоторых инфекционных болезней, таких как, например, инфекционные заболевания дыхательных путей, желудочнокишечного тракта, мочеполовых путей и наружных органов, таких как кожа, глаза и уши. Вследствие своей низкой стабильности в кислой среде эритромицин А необратимо превращается, например, в желудочно-кишечном тракте при оральном введении, в производное, лишенное антибиотической активности, что определяет плохую биологическую доступность действующего вещества Н. А. ,, 25,119-128 (1989). Для преодоления вышеуказанных недостатков исследование было направлено на соединения, которые, сохраняя хорошие антибиотические свойства эритромицина А, характеризовались бы более высокой стабильностью по отношению к кислотам и лучшими фармакокинетическими свойствами, такими как, например, более высокая биологическая доступность при оральном введении, концентрация в крови, способность проникать в ткани и период полураспада. В качестве примера в данной области техники можно привести карбаматы и карбонаты эритромицина А или 9-О-оксима эритромицина А, описанные в заявках на европейский патент 0216169 и 0284203 (обе на имя), а также соединения,описанные в европейской заявке 0033255 (фирма -). В европейской заявке 0033255, в частности, описаны 9 оксимовые производные эритромицина А формулы,где Еу обозначает остаток эритромицина А, в котором оксимовая группа (-) замещает карбонильную группу в положении 9 (Еу) А обозначает прямую или разветвленную алкильную группу с 1-6 атомами углеродаобозначает, в частности,необязательно замещенную алкоксигруппу с 1-6 атомами углерода или группу -(1)2,в которой 1 и 2 имеют одинаковые или различные значения и обозначают атом водорода или необязательно замещенную алкильную группу с 1-6 атомами углерода. Соединения, описанные в заявке на европейский патент 0033255, такие как, например,9- О- (2-метоксиэтокси)метил оксим эритромицина А, известный под международным незапатентованным названием рокситромицин,изд.,8253, 9-О(2-диметиламино)этилоксимэритромицина А и 9-О-(2-диэтиламино)этилоксимэритромицина А, обладают спектром активности, сопоставимым с таковым эритромицина А, но обладают более высокой стабильностью по отношению к кислотам и лучшими фармакокинетическими свойствами. Указанные соединения, следовательно, проявляют антибиотическую активность по отношению к грамположительным микроорганизмам,таким как стафилококки, стрептококки и пневмококки, и по отношению к некоторым грамотрицательным микроорганизмам, таким как, например,и. Были обнаружены соединения, являющиеся 9-О-оксимовыми производными эритромицина А, в частности соединения, представляющие собой производные 9-О-(аминоалкил)оксимаэритромицина А, частично подпадающие под объем, но не приведенные в качестве примера в европейской заявке 0033255, которые обладают более широким спект 3 4962 1 ром антибактериальной активности по отношению к грамположительным микроорганизмам, прежде всего по отношению к грамотрицательным микроорганизмам, и улучшенными фармакокинетическими свойствами, такими, как, например, бльшая продолжительность действия и больший период полураспада при элиминации в тканях по сравнению с соединениями, описанными в вышеуказанной европейской заявке. Таким образом, предметом изобретения являются 9-О-оксимовые производные эритромицина А общей формулы где А обозначает фенил, необязательно замещенный 1-3 одинаковыми или различными группами, выбранными из прямых или разветвленных С 1-С 4-алкокси, С 1-С 2-алкилендиокси, С 1-С 4-алкилсульфонила, фенила, фенокси, гидрокси, нитро, галогена и трифторметила, пиридил или фурил 1 и 2 одинаковые или различные, обозначают водород или прямой или разветвленный С 1-С 4-алкил- 1 или 2- целое число 1-8- целое число 2-6 М - группа формулы 3 3 где 3 - водород,или их фармацевтически приемлемые соли. Предпочтительно соединение общей формулыпо изобретению имеет Е-конфигурацию. Предпочтительными соединениями являются соединения общей формулы , где А пиридил, фурил или фенил, необязательно замещенный 1-3 группами, выбранными из гидрокси, метокси, метилендиокси, н-бутокси, фенокси, фенила, метилсульфонила, нитро,галогена и трифторметила 1 и 2 одинаковые или различные, обозначают водород или метил. Еще более предпочтительными соединениями являются соединения общей формулы ,где А обозначает фенил, необязательно замещенный группой, выбранной из фенокси,нитро и трифторметила 1 и 2 одинаковые и обозначают водород или метил- ,- 6, - 2. Другим объектом изобретения является фармацевтическая композиция, обладающая антибиотической активностью, содержащая терапевтически эффективное количество производного эритромицина А в смеси с фармацевтически приемлемым носителем. В качестве производного эритромицина А она содержит 9-О-оксимовое производное эритромицина А общей формулыпо изобретению. 4 4962 1 Соединения формулыпроявляют антибиотическую активность и характеризуются высокой стабильностью по отношению к кислотам и хорошими фармакокинетическими свойствами, что позволяет применять их в терапии человека или в ветеринарии для лечения некоторых инфекционных заболеваний, таких, как, например, заболевания центральной нервной системы, верхних и нижних дыхательных путей, желудочно-кишечного тракта,мочеполовых путей, зубной ткани и наружных органов, таких как кожа, глаза и уши. В настоящем описании, если не указано иное, под понятием (1-4) алкильная группа понимают прямую или разветвленную (1-4)алкильную группу, такую как метил, этил,н-пропил, изопропил, н-бутил, изобутил, втор-бутил и трет-бутил под понятием (14)алкоксигруппа понимают прямую или разветвленную (1-4)алкоксигруппу, такую,как метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси и трет-бутокси под понятием (С 1-С 2)алкилендиоксигруппа понимают метилендиокси- или этилендиоксигруппу. Фармацевтически приемлемые соли соединений формулыпредставляют собой соли органических или неорганических кислот таких как, например, соляная, бромистоводородная, йодистоводородная, азотная, серная, фосфорная, уксусная, винная, лимонная,бензойная, янтарная и глутаровая кислота. Конкретными примерами предпочтительных соединений формулыявляются(Е)-9-О-2 метил-6 метил(4-трифторметилфенил)метиламиногексиламиноэтилоксимэритромицина А. Получение соединений формулы , являющихся предметом настоящего изобретения,может быть осуществлено в соответствии с описанным ниже способом синтеза. Способ включает, во-первых, реакцию конденсации между соответствующей кислотой формулы 1(2)-1 где 1 иимеют указанные выше значения, с хлористым ацилом формулы А-(СН 2)-1-СОС,где А иимеют указанные выше значения. Реакцию конденсации осуществляют в соответствии со стандартными методами в инертном растворителе и в присутствии основания, такого как, например, гидроксид щелочного металла, с получением соединений формулы 1 где , 1,иимеют указанные выше значения. Полученные таким образом -ациламинокислоты формулыдалее подвергают конденсации в соответствии со стандартными методами с аминоэфирами формулы 2,)(2)-1 4 где 2 иимеют указанные выше значения,4 обозначает метильную или этильную группу, с получением соединений формулы 1 2,(2)-1(2)-1(2)-1 4 где , 1, 2, 4 ,иимеют указанные выше значения. В соответствии со стандартными методами соединения формулыдалее восстанавливают, например, с помощью борогидрида натрия в присутствии кислот, алюмогидрида лития, диметилсульфидборана или путем каталитического гидрирования, до соответствующих аминоспиртов формулы 1 2,(2)(2)(2)где , 1, 2, ,иимеют указанные выше значения. Аминоспирты формулызатем превращают в соответствующие сульфонильные производные формулы , например, с помощью метансульфонилхлорида или пара 6 4962 1 толуолсульфонилхлорида, и далее конденсируют с 9-О-оксимом эритромицина А или с 9 О-оксимом 6-О-метилэритромицина А, причем оба соединения могут быть представлены формулой , с получением соединений формулы 1 2(2)(2)(2)где , 1, 2, , ,иимеют указанные выше значения,5 обозначает мезильную или тозильную группу. Реакцию между соединениями формулыи оксимами формулыосуществляют в инертном органическом растворителе, таком как, например, тетрагидрофуран, этиловый эфир или 1,2-диметоксиэтан, в присутствии трет-бутилата калия и 18-краун-6 эфира в качестве комплексообразующего агента. Для специалиста в данной области техники очевидно, что, если реакцию сульфонилирования осуществляют с использованием соединений формулы , в которых один или оба заместителя 1 и 2 обозначают атом водорода, может оказаться необходимым защитить атом или атомы азота перед осуществлением реакции сульфонилирования. В этом случае конденсацию полученных таким образом -защищенных сульфонильных производных с оксимами формулыосуществляют аналогично тому, как это описано выше, а последующее удаление защитной группы осуществляют в соответствии с принятыми методами, что дает возможность получить соединения формулы , в которых один или оба заместителя 1 и 2 обозначают атом водорода. Более подробно защита аминов описана в литературе, например, см. и ,,, ., 2-е изд., (1991), 309-405. Соединения формул ,иявляются известными или легко могут быть получены в соответствии с известными методами. Оксимы формулытакже являются известными соединениями и могут быть получены в соответствии со стандартными методами, включающими, например, взаимодействие эритромицина А или 6-О-метилэритромицина А с гидрохлоридом гидроксиламина. Сложные эфиры формулынеобязательно могут быть получены в соответствии с альтернативным методом синтеза, включающим, во-первых, конденсацию соответствующей аминокислоты формулыс аминоэфиром формулыс получением соединений формулы 1 2(2)-1(2)-1 4 где 1, 2, 4,иимеют указанные выше значения. Специалисту в данной области техники очевидно, что перед осуществлением реакции конденсации между аминокислотой формулыи аминоэфиром формулыможет оказаться необходимым соответствующим образом защитить аминогруппу аминокислоты формулыаналогично тому, как это уже было описано для реакции сульфонилирования. Последующую конденсацию соединений формулыс соединением формулыосуществляют в соответствии со стандартными методами, а необязательное удаление защитной группы затем позволяет получить соединения формулы . Получение соединений формулы , в которых по крайней мере один из двух заместителей 1 и 2 обозначает группу, выбранную из этила, н-пропила, н-бутила и изобутила,7 4962 1 может быть осуществлено в соответствии с альтернативным способом синтеза, описанным ниже. Этот способ включает, во-первых, ацилирование атома или атомов азота аминоспиртов формулы , где один или оба заместителя 1 и 2 обозначают атом водорода. Например, при использовании соединения формулы , в котором оба заместителя 1 и 2 обозначают атом водорода, в соответствии со стандартными методами в присутствии пригодного ацилхлоридаможно получить соединения формулы где , ,иимеют указанные выше значения,обозначает прямую или разветвленную (С 1-С 3)алкильную группу. Восстановление соединений формулы , осуществляемое в соответствии со стандартными методами, позволяет получить соединения формулы 2 1(2), (2) где , ,иимеют указанные выше значения,1 и 2 обозначают этил, н-пропил, нбутил или изобутил, из которых после превращения в соответствующие сульфонильные производные и конденсации с оксимами формулыаналогично тому, как это описано выше, можно получить соединения формулы 1 2(2)(2)(2),где , , ,иимеют указанные выше значения,1 и 2 обозначают этил, н-пропил,н-бутил или изобутил. Ниже описан процесс синтеза, альтернативный по отношению к тому, который описан ранее для получения соединений формулы , являющихся предметом настоящего изобретения. Этот процесс включает, во-первых, окисление соответствующего -защищенного аминоспирта, такого как, например, -бензилоксикарбониламиноспирт формулы , в присутствии гипохлорита натрия и свободного радикала 2,2,6,6-тетраметилпиперидинокси(ТЕМПО) в инертном органическом растворителе с получением соединений формулы гдеимеет указанные выше значения,обозначает защитную группу. Примерами инертных органических растворителей, пригодных для реакции окисления,являются, например, метиленхлорид, хлороформ, черыреххлористый углерод, 1,2-дихлорэтан, этилацетат, бензол и толуол. Аминирование полученного таким образом альдегида в присутствии пригодного аминоспирта формулыи восстановление полученного промежуточного продукта, например, в присутствии боргидрида натрия, позволяет получить аминоспирты формулы где ,иимеют указанные выше значения.,4962 1 Последующая защита азота аминогруппы соединений формулыи тем самым превращение в соответствующие сульфонильные производные, конденсация с оксимами формулыи удаление защитной группы атомов азота аналогично тому, как это описано выше, позволяют получить соединения формулы где ,иимеют указанные выше значения. Промежуточные оксимы формулы , сконденсированные с соответствующим альдегидом формулыи восстановленные, например, каталитическим гидрированием, позволяют получить соединения формулы где А, М,иимеют указанные выше значения. Соединения формул ,иявляются известными или легко могут быть получены в соответствии с известными методами. Соединения формулы , в которых один или оба заместителя 1 и 2 обозначают атом водорода, полученные в соответствии с одним из описанных выше способов, необязательно могут быть алкилированы по атому или атомам азота диаминофрагмента в соответствии со стандартными методами, включающими, например, конденсацию с соответствующим альдегидом и восстановление полученного промежуточного продукта. Таким путем получают соединения формулы , в которых 1 и 2 имеют одинаковые или различные значения и обозначают прямую или разветвленную (1-С 4)алкильную группу. Получение соединений формулы , имеющих - или Е-кофигурацию, осуществляют в соответствии с одной из схем синтеза, описанных выше, используя оксим формулыв требуемой конфигурации и др.,, 44, 313-330, (1991). Было установлено, что соединения формулыпроявляют антибактериальную активность по отношению к некоторым грамположительным и грамотрицательным микроорганизмам и пригодны для применения в клинической терапии и в ветеринарии при лечении некоторых инфекционных заболеваний, таких как, например, заболевания центральной нервной системы, верхних и нижних дыхательных путей, желудочно-кишечного тракта,мочеполовых путей, зубной ткани и наружных органов, таких как кожа, глаза и уши. Кроме того, эти соединения обладают активностью по отношению к некоторым представляющим клинический интерес грамположительным микроорганизмам, устойчивым к эритромицину А или в более общем случае к антибиотикам из класса макролидов, характеризующихся наличием 14- или 15-членных макролактонов. Антибактериальную активность соединений формулыпо отношению к грамположительным микроорганизмам, таким как,,и, и грамотрицательным микроорганизмам, таким каки, оценивали с помощью опытов,пригодных для оценки минимальной концентрации антибиотика, позволяющей ингибировать рост бактерий (МИК) (пример 23). В качестве эталонов применяли рокситромицин и кларитромицин,изд.,8253 и 2340 соответственно. Было установлено, что антибактериальная активность соединений формулыпо отношению к грамположительным микроорганизмам практически сопоставима с таковой рокситромицина и кларитромицина, т.е. двух макролидов, характеризующихся в опытахвысокой антибактериальной активностью (таблица 1). Было установлено также, что по отношению к грамотрицательным микроорганизмам,в частности по отношению к энтеробактериям, таким каки 9, соединения формулыявляются существенно более активными по сравнению с обоими соединениями, используемыми в качестве эталонов (таблица 2). В этой связи интересно отметить, что соединения формулы , являющиеся предметом настоящего изобретения, оказались более активными, чем рокситромицин, описанный в вышеуказанной европейской заявке 0033255 и выбранный в качестве предпочтительного соединения по отношению к нескольким другим производным, таким как, например, 9 О- (2-диметиламино)этилоксим эритромицинаи др.,, 44, 313-330 (1991). Кроме того, было установлено, что соединения формулыпроявляют активность в опытах(таблица 3). Антибактериальную активность соединений формулыв опытах, выраженную в виде средней защищающей дозы 50 (мг/кг), оценивали с помощью экспериментальной легочной инфекции, вызванной у мышей с помощью(пример 23). При анализе данных об активности, обнаруженной в опытах, становится очевидным, что соединения формулыотличаются пролонгированным действием и длительным периодом полураспада при элиминации из ткани. Действительно, после внутрибрюшинного введения мыши соединения формулыбыстро и повсеместно распределялись по всему организму, а уровни в тканях оказывались выше, чем в плазме. Эти результаты становятся особенно очевидными при анализе значений 50 для соединений формулы , введенных за 24 часа до или через 1 час после заражения. Действительно, эти величины оказались практически неизмененными после введения за 24 часа до или через 1 час после заражения. В случае экспериментальной легочной инфекции, вызванной у мышей с помощью, т.е. патогена, обычно вызывающего респираторные заболевания,эффективные концентрации соединений формулы , введенные внутрибрюшинно, сохранялись в области легких в течение 24-48 часов после введения. В отличие от этого рокситромицин и кларитромицин, т.е. используемые в качестве эталонов соединения, будучи введенными за 24 часа до заражения, оказались неактивными. Следовательно, соединения формулытакже обладают избирательной активностью по отношению к легким и могут успешно применяться для лечения инфекций дыхательных путей. В дополнение к вышеуказанной активности по отношению к бактериальным микроорганизмам соединения формулы , являющиеся предметом настоящего изобретения, являются активными по отношению к эукариотным патогенам, в частности, они обладают выраженной активностью по отношению к простейшим, таким как,который является хорошо известным возбудителем малярии. Следовательно, соединения формулытакже могут успешно применяться для лечения заболеваний малярией. Наряду с тем, что они характеризуются широким спектром антибиотической активности по отношению к грамположительным и грамотрицательным микроорганизмам и простейшим, хорошей стабильностью по отношению к кислотам и хорошими фармакокинетическими свойствами, острая токсичность для мышей соединений формулысопоставима с таковой рокситромицина. Следовательно, отличаясь высокой безопасностью при применении, они могут успешно использоваться в терапии человека и в ветеринарии. Соединения формулыпредпочтительно следует использовать в соответствующей фармацевтической форме, пригодной для орального, парентерального введения, в виде суппозитория или для местного применения. Таким образом, предметом настоящего изобретения являются фармацевтические композиции, содержащие терапевтически эффективное количество одного или нескольких соединений формулыв смеси с фармацевтически приемлемым носителем. Эти фармацевтические формы включают таблетки, капсулы, сиропы, растворы для инъекций, готовые к применению или получаемые перед применением путем разбавления лиофилизированной формы, суппозитории, растворы, кремы, мази и глазные примочки. Для ветеринарных целей в дополнение к вышеуказанным композициям 10 4962 1 возможно изготовление твердых или жидких концентратов, подлежащих разбавлению в пище или в воде для питья. В соответствии с типом композиции кроме терапевтически эффективного количества одного или нескольких соединений формулыэти композиции должны содержать твердые или жидкие эксципиенты или растворители для фармацевтической или ветеринарной целей и необязательно другие добавки, обычно используемые в технике изготовления препаративных форм, такие как загущающие агенты, агрегирующие агенты, замасливатели, агенты, способствующие дезинтеграции, корригенты и красители. Для лечения конкретных инфекций соединение формулыможет быть объединено с эффективным количеством другого действующего вещества. Эффективное количество соединения формулыможет варьироваться в зависимости от различных факторов, таких как серьезность и фаза заболевания, орган или система,подверженная заболеванию, характеристики видов хозяев, чувствительность бактериальных видов, обусловливающих инфекцию, и выбранный путь введения. Терапевтическая доза обычно составляет от 0,5 до 100 мг/кг веса тела в день и может применяться в виде однократной дозы или нескольких суточных доз. Ниже изобретение проиллюстрировано на примерах, не ограничивающих его объем. Строение соединений формулыи промежуточных продуктов синтеза при их получении подтверждали с помощью 1 Н-ЯМР- или 13 С-ЯМР-спектроскопии. Величины значимых сигналов наиболее важных промежуточных продуктов и соединений формулыприведены ниже в описании. Пример 1. Получение -бензоил-6-аминокапроновой кислоты Раствор бензоилхлорида (0,18 моля) в этиловом эфире (160 мл) и раствор 1 н. гидроксида натрия (180 мл) одновременно добавляли к смеси 6-аминокапроновой кислоты (0,15 моля) в этиловом эфире (150 мл) и воды (200 мл), содержащей гидроксид натрия (0,15 моля), и перемешивали при температуре от 0 до 5 С. По завершении добавления соединений температуру реакционной смеси доводили до комнатной и перемешивали в течение еще 4 часов. После разделения фаз водную фазу промывали этиловым эфиром (200 мл) и подкисляли концентрированной соляной кислотой с использованием реактивной бумаги конго. После экстракции этилацетатом (3200 мл) объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (200 мл), сушили над сульфатом натрия и упаривали при пониженном давлении. Таким путем получали -бензоил-6-аминокапроновую кислоту, которую непосредственно применяли в последующих реакциях. По аналогичной методике получали следующие соединения-бензоилизопропил-6-аминокапроновую кислоту. Пример 2. Получение этилового эфира -6-(бензоиламино)гексаноил глицина Раствор дициклогексилкарбодиимида (112 ммолей) в безводном тетрагидрофуране(44 мл) постепенно добавляли к суспензии, содержащей -бензоил-6-аминокапроновую кислоту (93,5 ммоля), полученную согласно примеру 1, гидрохлорид этилового эфира глицина (112 ммолей), триэтиламин (112 ммолей) и безводный 1-гидроксибензотриазол(112 ммолей) в тетрагидрофуране (330 мл) и перемешивали при температуре 0 С. Далее температуру реакционной смеси доводили до комнатной и перемешивали в течение 16 часов. 11 4962 1 В результате получали осадок, который удаляли фильтрацией, а полученный таким образом фильтрат упаривали при пониженном давлении. Остаток объединяли с этилацетатом(300 мл) и последовательно промывали раствором 5 -ной соляной кислоты (2100 мл),насыщенным раствором хлорида натрия (100 мл), раствором 5 -ного бикарбоната натрия(2100 мл) и в завершение насыщенным раствором хлорида натрия (100 мл). Органическую фазу сушили над сульфатом натрия и упаривали досуха при пониженном давлении,получая таким образом этиловый эфир -6-(бензоиламино)гексаноилглицина, который непосредственно применяли в последующих реакциях. По аналогичной методике получали следующие соединения этиловый эфир -(бензоиламино)ацетилглицина этиловый эфир -6-(фенилацетиламино)гексаноилглицина этиловый эфир -(фенилацетиламино)ацетилглицина этил-6-6-(бензоиламино)гексаноиламиногексаноат метиловый эфир -5-(бензоиламино)пентаноилглицина метил-6-5-(бензоиламино)пентаноиламиногексаноат метиловый эфир -7-(бензоиламино)гептаноилглицина метил-5-6-(бензоиламино)гексаноиламинопентаноат метил-6-(бензоиламино)ацетиламиногексаноат метил-3-6-(бензоиламино)гексаноиламинопропионоат этил-6 изопропил(фенилацетиламино)ацетиламиногексаноат метил-6-4-(бензоиламино)бутаноиламиногексаноат метил-4 изопропил-4-(-изопропилбензоиламино)бутаноил-аминобутаноат. Пример 3. Получение этилового эфира -(6-аминоногексаноил)глицина а) К раствору гидроксида натрия (33,54 г 0,831 моля) в воде (840 мл) и метаноле (400 мл) добавляли 6-аминокапроновую кислоту (100 г 0,762 моля) и постепенно добавляли раствор ди-трет-бутилдикарбоната (168 г 0,762 моля) в метаноле (140 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. После этого вновь добавляли твердый ди-трет-бутилдикарбонат (17,5 г 0,08 моля) и реакционную смесь перемешивали еще в течение 16 часов. Затем реакционную смесь промывали гексаном (2400 мл),подкисляли раствором бисульфата калия до рН 1,5 и экстрагировали этилацетатом (3450 мл). Объединенные органические фазы сушили над сульфатом натрия и упаривали досуха, получая при этом 6-(трет-бутоксикарбониламино) капроновую кислоту (163 г) в виде масла. б) Аналогично методике, описанной в примере 2, 6-(трет-бутоксикарбониламино)капроновую кислоту (163 г) непосредственно конденсировали с гидрохлоридом этилового эфира глицина (118 г 0,845 моля), получая таким образом этиловый эфир -6-(трет-бутоксикарбониламино)гексаноилглицина (285 г) в виде неочищенного продукта, который непосредственно применяли в последующей реакции, пл 76-77 С (изопропиловый эфир). в) Раствор 6 н. соляной кислоты (150 мл) в этилацетате (150 мл) добавляли к раствору этилового эфира -6-(трет-бутоксикарбониламино)гексаноилглицина (285 г) в этилацетате (500 мл) и перемешивали при комнатной температуре. Через 24 часа образовывался осадок, который отфильтровывали, промывали этилацетатом и этиловым эфиром и сушили в термостате (50 С) под вакуумом. Таким путем получали этиловый эфир -(6-аминоногексаноил)глицина (93 г) в виде неочищенного продукта, который непосредственно применяли в последующих реакциях. ТСХ (метиленхлоридметаноламмиак 1021)0,2. Пример 4. Получение этилового эфира -6-(4-фторбензоил)аминогексаноилглицина Раствор 4-фторбензоилхлорида (47,4 ммоля) в метиленхлориде (30 мл) постепенно добавляли к суспензии, содержащей этиловый эфир -(6-аминоногексаноил)глицина (39,5 ммоля), полученный согласно примеру 3, и триэтиламин (87 ммолей) в метиленхлориде 12(150 мл) и перемешивали при 0 С. Температуру полученной таким образом смеси, к которой затем добавляли триэтиламин (2 мл), доводили до комнатной и перемешивали. После выдержки в течение 1 часа в этих условиях реакционную смесь промывали раствором 5 -ной соляной кислоты (2100 мл) и насыщенным раствором хлорида натрия (3100 мл). Отделенную органическую фазу сушили над сульфатом натрия и упаривали досуха под вакуумом. Таким путем получали этиловый эфир -6-(4-фторбензоил)аминогексаноилглицина в виде неочищенного продукта, который непосредственно применяли в последующих реакциях, пл 121-122 С (этилацетат) ТСХ (этилацетат)0,3. По аналогичной методике получали следующее соединениеэтиловый эфир -6-(2-фуроиламино)гексаноил глицина, пл 104-106 С (ацетонитрил/изопропиловый эфир) ТСХ (метиленхлоридметанол 955)0,3. Пример 5. Получение этилового эфира -6-(4-метоксибензоил)аминогексаноилглицина Аналогично примеру 2 с использованием 4-метоксибензойной кислоты (33 ммоля) и этилового эфира -(6-аминоногексаноил)глицина (39,5 ммоля), полученного согласно примеру 3, получали этиловый эфир -6-(4-метоксибензоил)аминогексаноилглицина в виде неочищенного продукта, который непосредственно применяли в последующих реакциях, пл 106-107 С. ТСХ (метиленхлоридметанол 9010)0,46. По аналогичной методике получали следующие соединенияэтиловый эфир -6-(3,4-метилендиоксибензоил)аминогексаноилглицина, ТСХ (метиленхлоридметанол 9010)0,39 этиловый эфир -6-(4-метилсульфонилбензоил)аминогексаноилглицина, пл 124126 С ТСХ (метиленхлоридметанол 964)0,31 этиловый эфир -6-(3-трифторметилбензоил)аминогексаноилглицина, пл 102104 С ТСХ (метиленхлоридметанол 955)0,38. Пример 6. Получение 2-6-(фенилметиламино)гексиламиноэтанола Серную кислоту (6 н.) в этиловом эфире (40,9 мл 700 ммолей), полученную смешением 96 -ной серной кислоты (33 мл) и этилового эфира (100 мл), постепенно добавляли к суспензии, содержащей этиловый эфир -6-(бензоиламино)гексаноилглицина (46,8 ммоля), полученный согласно примеру 2, и борогидрид натрия (700 ммолей) в безводном тетрагидрофуране (200 мл), и перемешивали при температуре от 15 С до 20 С. Реакционную смесь доводили до температуры кипения в течение 24 часов и затем охлаждали до 0 С. Затем при перемешивании добавляли метанол (150 мл). Растворитель выпаривали при пониженном давлении и остаток объединяли с раствором 6 н. гидроксида натрия (200 мл), выдерживая полученную смесь при температуре кипения в течение 24 часов. Затем реакционную смесь, охлажденную до комнатной температуры, экстрагировали тетрагидрофураном (2100 мл) и органическую фазу упаривали досуха, объединяли с этилацетатом и сушили над сульфатом натрия. Путем подкисления эфирным раствором соляной кислоты получали осадок, представляющий собой 2-6-(фенилметиламино)гексиламино этанол в виде гидрохлорида. Полученный таким образом неочищенный продукт непосредственно применяли в последующих реакциях. По аналогичной методике получали следующие соединения 2-2-(фенилметиламино)этиламиноэтанол 2-6-(2-фенилэтиламино)гексиламиноэтанол 6-6-(фенилметиламино)гексиламиногексанол 2-5-(фенилметиламино)пентиламиноэтанол 2-8-(фенилметиламино)октиламиноэтанол 5-6-(фенилметиламино)гексиламинопентанол 6-3-(фенилметиламино)пропиламиногексанол 3-6-(фенилметиламино)гексиламинопропанол 13 4962 1 3-4-(фенилметиламино)бутиламинопропанол 6-2-(фенилметиламино)этиламиногексанол 6 изопропил-4-(фенилметиламино)бутиламиногексанол 2-6-(4-фторфенил)метиламиногексиламиноэтанол 2-6-(4-метоксифенил)метиламиногексиламиноэтанол 2-6-(3,4-метилендиоксифенил)метиламиногексиламиноэтанол 2-6-(3-трифторметилфенил)метиламиногексиламиноэтанол 2-6-(4-метилсульфонилфенил)метиламиногексиламиноэтанол 4 изопропил-4-(-изопропилфенилметиламино)бутиламинобутанол. Пример 7. Получение 6 ацетил-6-(-ацетилфенилметиламино)гексиламиногексилацетата Триэтиламин (1,95 мл 14 ммолей) и раствор ацетилхлорида (0,62 мл 8,69 ммоля) в метиленхлориде (5 мл) постепенно добавляли к суспензии, содержащей 6-6-(фенилметиламино)гексиламиногексанол (1 г 2,6 ммоля), полученный согласно примеру 6, в метиленхлориде (15 мл), и перемешивали при 0 С. После перемешивания в течение 1 часа при 0 С температуру реакционной смеси доводили до комнатной и перемешивали еще в течение 16 часов. Затем реакционную смесь промывали раствором 10 -ной соляной кислоты(10 мл) и насыщенным раствором хлорида натрия. После разделения фаз органическую фазу сушили над сульфатом натрия и упаривали досуха под вакуумом, получая таким образом 6 ацетил-6-(-ацетилфенилметиламино)гексиламиногексилацетат (1,18 г) в виде масла, который непосредственно применяли в последующих реакциях. По аналогичной методике получали следующее соединение 2 ацетил-6 ацетил(2-фенилэтил)аминогексиламиноэтилацетат. Пример 8. Получение 6 этил-6-(-этилфенилметиламино)гексиламиногексанола Аналогично примеру 6 с использованием 6 ацетил-6-(-ацетилфенилметиламино)гексиламиногексилацетата, полученного согласно примеру 7, получали 6 этил 6-(-этилфенилметиламино)гексиламиногексанол. По аналогичной методике получали следующее соединение 2 этил-6 этил(2-фенилэтил)аминогексиламиноэтанол. Пример 9. Получение 2 бензилоксикарбонил-6-(-бензилоксикарбонилфенилметиламино)гексиламиноэтанола Раствор 1 н. гидроксида натрия (44,5 мл) и толуоловый раствор 50 -ного бензилхлорформиата (44,5 ммоля) в этилацетате (33 мл) постепенно и одновременно добавляли к раствору дигидрохлорида 2-6-(фенилметиламино)гексиламиноэтанола (18,5 ммоля), полученного согласно примеру 6, в растворе 1 н. гидроксида натрия (37,1 мл) и этилацетата(40 мл), и перемешивали при температуре 0 С. По завершении добавлений температуру реакционной смеси доводили до комнатной и перемешивали в течение 24 часов. После разделения фаз водную фазу промывали этилацетатом (250 мл). Объединенные органические фазы промывали насыщенным раствором хлорида натрия (50 мл), сушили над сульфатом натрия и упаривали досуха под вакуумом. Таким путем получали 2 бензилоксикарбонил-6-(-бензилоксикарбонилфенилметиламино)гексиламино этанол в виде масла, который непосредственно применяли в последующих реакциях. ТСХ (этилацетатгексан 5050)0,20. По аналогичной методике получали следующие соединения 2 бензилоксикарбонил-2-(-бензилоксикарбонилфенилметиламино)этиламиноэтанол, ТСХ (этилацетатгексан 6040)0,25 6 бензилоксикарбонил-6-(-бензилоксикарбонилфенилметиламино)гексиламиногексанол, ТСХ (этилацетатгексан 5050)0,27 6 бензилоксикарбонил-5-(-бензилоксикарбонилфенилметиламино)пентиламиногексанол 14 4962 1 2 бензилоксикарбонил-5-(-бензилоксикарбонилфенилметиламино)пентиламиноэтанол 2 бензилоксикарбонил-8-(-бензилоксикарбонилфенилметиламино)октиламиноэтанол 5 бензилоксикарбонил-6-(-бензилоксикарбонилфенилметиламино)гексиламинопентанол 6 бензилоксикарбонил-3-(-бензилоксикарбонилфенилметиламино)пропиламиногексанол 3 бензилоксикарбонил-6-(-бензилоксикарбонилфенилметиламино)гексиламинопропанол 3 бензилоксикарбонил-4-(-бензилоксикарбонилфенилметиламино)бутиламинопропанол 6 изопропил-2 бензилоксикарбонил(2-фенилэтил)аминоэтиламиногексанол,ТСХ (метиленхлоридметаноламмиак 9550,5)0,33 6 бензилоксикарбонил-4-(-изопропилфенилметиламино)бутиламиногексанол,ТСХ (метиленхлоридметаноламмиак 9550,5)0,42 2 бензилоксикарбонил-6 бензилоксикарбонил(4-фторфенил)метиламиногексиламиноэтанол, ТСХ (этилацетатгексан 6040)0,35 2 бензилоксикарбонил-6 бензилоксикарбонил(4-метоксифенил)метиламиногексиламиноэтанол, ТСХ (этилацетатгексан 5050)0,2 2 бензилоксикарбонил-6 бензилоксикарбонил(3,4-метилендиоксифенил)метиламиногексиламиноэтанол, ТСХ (этилацетатгексан 6040)0,26 2 бензилоксикарбонил-6 бензилоксикарбонил(3-трифторметилфенил)метиламиногексиламиноэтанол, ТСХ (этилацетатгексан 5050)0,25 2 бензилоксикарбонил-6 бензилоксикарбонил(4-метилсульфонилфенил)метиламиногексиламиноэтанол, ТСХ (этилацетатгексан 9010)0,36. Пример 10. Получение 2 бензилоксикарбонил-6 бензилоксикарбонилфенилметиламино)гексиламиноэтилметансульфоната Раствор метансульфонилхлорида (3,16 ммоля) в метиленхлориде (5 мл) постепенно добавляли к раствору 2 бензилоксикарбонил-6-(-бензилоксикарбонилфенилметиламино)гексиламиноэтанола (2,6 ммоля), полученного согласно примеру 9, в метиленхлориде (15 мл), содержащем триэтиламин (0,44 мл 3,16 ммоля), и перемешивали при температуре 0 С. Реакционную смесь, температуру которой доводили до комнатной и перемешивали в течение 5 часов, добавляли к раствору 5 -ной соляной кислоты (20 мл). После разделения фаз органическую фазу промывали 5 -ной соляной кислотой (10 мл) и насыщенным раствором хлорида натрия (310 мл). Затем органическую фазу сушили над сульфатом натрия и упаривали досуха, получая таким образом 2 бензилоксикарбонил 6-(-бензилоксикарбонилфенилметиламино)гексиламиноэтилметансульфонат, который непосредственно применяли в реакции, приведенной в следующем примере. Пример 11. Получение (Е)-9-О-2 бензилоксикарбонил-6-(-бензилоксикарбонилфенилметиламино)гексиламиноэтилоксимаэритромицина А(Е)-9-О-оксим эритромицина А (627 мг 0,84 ммоля), 18-краун-6-эфир (220 мг 0,84 ммоля) и раствор 2 бензилоксикарбонил-6-(-бензилоксикарбонилфенилметиламино)гексиламиноэтилметансульфоната (0,84 ммоля), полученного согласно примеру 10, в безводном тетрагидрофуране (5 мл) добавляли в указанном порядке к суспензии трет-бутилата калия (103 мг 0,92 ммоля) в безводном тетрагидрофуране (5 мл) и выдерживали при комнатной температуре и при перемешивании в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение 20 часов и затем упа 15 4962 1 ривали при пониженном давлении. Остаток объединяли с этилацетатом (10 мл) и полученную таким образом смесь промывали насыщенным раствором хлорида натрия (10 мл). Водную фазу экстрагировали этилацетатом (210 мл) и объединенные органические фазы сушили над сульфатом натрия и упаривали досуха. Таким путем получали (Е)-9-О-2-бензилоксикарбонил-6-(-бензилоксикарбонилфенилметиламино)гексиламиноэтилоксимэритромицина А и применяли его непосредственно в последующих реакциях. ТСХ(метиленхлоридметаноламмиак 9091)0,58 (МН)1250 1 НЯМР (200 МГц, 3)(част./млн) 7,38-7,10 (, 15, ароматические соединения) 5,185,10 (, 4, 2 2) 3,30 (, 3 Н, ОСН 3) 2,26 (, 6, 2 3) 0,81 (, 3 Н, СН 3 СН 2). По аналогичной методике получали следующие соединения(, 2, 2) 3,30 (, 3 Н, ОСН 3) 2,26 (, 6, 23) 0,82 (, 3 Н, СН 3 СН 2). Пример 12. Получение (Е)-9-О-2-6-(фенилметиламино)гексиламиноэтилоксимаэритромицина А (соединение 4) К раствору (Е)-9-О-2 бензилоксикарбонил-6 бензилоксикарбонилфенилметиламино)гексиламиноэтилоксимаэритромицина А (5,9 ммоля), полученного согласно примеру 11, в этаноле (150 мл) добавляли 10 -ный палладий на угле (750 мг). Полученную таким образом смесь помещали в гидрогенизатор Парра, загруженный водородом(1 бар), и перемешивали при комнатной температуре. Через 7 часов катализатор отфильтровывали и спиртовой раствор упаривали досуха. Таким путем после очистки с помощью хроматографии на силикагеле (элюент метиленхлоридметаноламмиак 90101) получали (Е)-9-О-2-6-(фенилметиламино)гексиламиноэтилоксимэритромицина А. МС(МН)982 13 С-ЯМР (50 МГц 3)(част./млн) 140,48 128,39 128,11 126,88. По аналогичной методике получали следующие соединения(Е)-9-О-2-6-(4-метилсульфонилфенил)метиламиногексиламиноэтилоксим эритромицина А (соединение 20), МС (МН)1059 13 С-ЯМР (50 МГц, 3)(част./млн) 147,23 138,97 128,79 127,49. Пример 13. Получение -бензилоксикарбонил-6-аминогексанола Бензилхлорформиат (50 -ный в толуоле 84,8 мл 0,256 моля) в этилацетате (171 мл) и раствор 1 н. гидроксида натрия (256 мл) постепенно и одновременно добавляли к смеси 6-аминогексанола (25 г 0,21 моля) в этилацетате (250 мл) и воды (200 мл) и перемешивали при 0 С. Температуру реакционной смеси (рН 9) доводили до комнатной и перемешивали в течение 5 часов. После разделения фаз водную фазу промывали этилацетатом (200 мл). Затем объединенные органические фазы промывали насыщенным раствором хлорида на 18 4962 1 трия (150 мл), сушили над сульфатом натрия и упаривали досуха. Остаток объединяли с этиловым эфиром (300 мл) и образовавшийся осадок отфильтровывали и сушили под вакуумом при 50 С, получая таким образом -бензилоксикарбонил-6-аминогексанол (44,5 г),пл 80-82 С. Пример 14. Получение -бензилоксикарбонил-6-аминогексанала Раствор бромида калия (1,89 г 16 ммолей) в воде (31 мл) добавляли к раствору бензилоксикарбонил-6-аминогексанола (40 г 0,159 моля), полученного согласно примеру 13,в метиленхлориде (600 мл), содержащему свободный радикал 2,2,6,6-тетраметилпиперидинокси (ТЕМПО) (0,248 г 1,6 ммоля). К реакционной смеси для установления значения рН на 8,7 постепенно добавляли раствор гипохлорита натрия (215 мл), полученный путем смешения 7 -ного раствора гипохлорита натрия (240 мл) с бикарбонатом натрия (4,22 г) и 5 -ной соляной кислотой (5 мл), и перемешивали при 10 С. По окончании добавления и после разделения фаз органическую фазу промывали метиленхлоридом(2200 мл), сушили над сульфатом натрия и упаривали досуха. Таким путем получали бензилоксикарбонил-6-аминогексанал (39,45 г) в виде масла. ТСХ (этилацетатгексан 11)0,41. Пример 15. Получение 2-6-(бензилоксикарбониламино)гексиламиноэтанола Смесь, содержащую -бензилоксикарбонил-6-аминогексанал (35 г 0,14 моля) и 2 аминоэтанол (51,3 г 0,84 моля) в этаноле (250 мл), в присутствии молекулярных сит (3 ) перемешивали при комнатной температуре в течение 2 часов. Затем реакционную смесь фильтровали через целит и к образовавшемуся раствору добавляли борогидрид натрия(6,33 г 0,168 моля). После перемешивания в течение 4 часов при комнатной температуре реакционный растворитель выпаривали под вакуумом и остаток объединяли с водой (500 мл) и этилацетатом (500 мл). После разделения фаз водную фазу дополнительно экстрагировали этилацетатом (200 мл). Объединенные органические фазы промывали насыщенным раствором хлорида натрия (250 мл), сушили над сульфатом натрия и упаривали досуха,получая таким образом 2-6-(бензилоксикарбониламино)гексиламиноэтанол (38,36 г). ТСХ (этилацетатметаноламмиак 1021)0,4. Пример 16. Получение 2 бензилоксикарбонил-6-(бензилоксикарбониламино)гексиламиноэтанола Аналогично примеру 9 с использованием 2-6-(бензилоксикарбониламино)гексиламиноэтанола (38,3 г 0,13 моля), полученного согласно примеру 15, получали 2-бензилоксикарбонил-6-(бензилоксикарбониламино)гексиламиноэтанол в виде масла. ТСХ(этилацетатгексан 6535)0,45. Пример 17. Получение 2 бензилоксикарбонил-6-(бензилоксикарбониламино)гексиламино этилметансульфоната Аналогично примеру 10 с использованием 2 бензилоксикарбонил-6-(бензилоксикарбониламино)гексиламино этанола (20 г 47,8 ммоля), полученного согласно примеру 16,получали 2 бензилоксикарбонил-6-(бензилоксикарониламино)гексиламиноэтилметансульфонат (24,35 г), который использовали непосредственно в последующих реакциях. Пример 18. Получение (Е)-9-О-2 бензилоксикарбонил-6-(бензилоксикарбониламино)гексиламиноэтилоксимаэритромицина А Аналогично примеру 11 с использованием 2 бензилоксикарбонил-6-(бензилоксикарбониламино)гексиламиноэтилметансульфоната (24,25 г 47,8 ммоля), полученного согласно примеру 17, после хроматографии на силикагеле (элюент метиленхлоридметаноламмиак 9550,5) получали(, 3 Н, ОСН 3) 2,25 (, 6, 2 3) 0,80 (, 3 Н, СН 3 СН 2). Пример 19. Получение (Е)-9-О-2-(6-аминогексиламино)этилоксимаэритромицина А Аналогично примеру 12 с использованием (Е)-9-О-2 бензилоксикарбонил-6-(бензилоксикарбониламино)гексиламиноэтилоксимаэритромицина А, полученного согласно примеру 18, после хроматографии на силикагеле (элюент метиленхлоридметаноламмиак 85151,5) получали (Е)-9-О-2-(6-аминогексиламино)этилоксимэритромицина А. ТСХ (метиленхлоридметаноламмиак 85151,5)0,2 13 С-ЯМР (50 МГц, 3)(част./млн) 175,18 171,26 102,96 96,28. Пример 20. Получение (Е)-9-О-2-6-(2-трифторметилфенил)метиламиногексиламиноэтилоксимаэритромицина А (соединение 21) К раствору (Е)-9-О-2-(6-аминогексиламино)этилоксимаэритромицина А (2 г 2,24 ммоля), полученного согласно примеру 19, в этаноле (50 мл) добавляли 2 трифторметилбензальдегид (0,4 г) и молекулярные сита (4,5 г 3 ) и перемешивали при комнатной температуре. Через 2 часа молекулярные сита отфильтровывали и к образовавшемуся раствору добавляли 10 -ный палладий на угле (0,2 г). Реакционную смесь помещали в гидрогенизатор Парра, загруженный водородом (1 бар). Через 1 час по окончании реакции гидрирования катализатор отфильтровывали и растворитель выпаривали. Остаток очищали с помощью хроматографии на силикагеле (элюент метиленхлоридметаноламмиак 9550,5), получая таким образом (Е)-9-О-2-6-(2-трифторметилфенил)метиламиногексиламиноэтилоксимэритромицина А (2 г). МС (МН)1050 13-ЯМР (50 МГц, 3)(част./млн) 139,14 131,88 130,38 127,58 126,81 125,82. По аналогичной методике получали следующие соединения(2,5 г 2,8 ммоля), полученного согласно примеру 19, в безводном этаноле (100 мл). Реакционную смесь перемешивали при комнатной температуре и через 2 часа молекулярные сита отфильтровывали и к образовавшемуся раствору порциями добавляли борогидрид натрия (0,106 г 2,89 ммоля). После перемешивания в течение 3 часов растворитель выпаривали при пониженном давлении и остаток очищали с помощью хроматографии на силикагеле (элюент метиленхлоридметаноламмиак 85151,5), получая таким образом(Е)-9-О-2-6-(3,5-дихлор-2-гидроксифенил)метиламиногексиламиноэтилоксимэритромицина А (2,2 г). МС (МН)1066 13 С-ЯМР (50 МГц, 3)(част./млн) 153,43 128,43 126,42 124,41 122,91 121,61. По аналогичной методике получали следующие соединения(част./млн) 155,50 151,98 132,51 125,13 119,67 118,59. Пример 22. Получение (Е)-9-О-2 метил-6-(-метилфенилметиламино)гексиламиноэтилоксимаэритромицина А (соединение 38) Водный 37 -ный раствор формальдегида (2 мл 26,6 ммоля) и 10 -ный палладий на угле (0,82 г) добавляли в указанной последовательности к раствору (Е)-9-О-2-6(фенилметиламино)гексиламиноэтилоксимаэритромицина А (2 г 2 ммоля), полученного согласно примеру 12, в смеси этанолвода 11 (20 мл) и перемешивали при комнатной температуре. Реакционную смесь помещали в гидрогенизатор Парра, загруженный водородом (1 бар). Через 2 часа реакционную смесь фильтровали для удаления катализатора и образовавшийся раствор упаривали досуха. Образовавшийся остаток очищали с помощью хроматографии на силикагеле (элюент метиленхлоридметаноламмиак 90101),получая (Е)-9-О-2 метил-6-(-метилфенилметиламино)гексиламиноэтилоксимэритромицина А (1,8 г). МС (МН)1009 13 С-ЯМР (50 МГц, 3)(част./млн) 139,20 129,04 128,17 126,86. По аналогичной методике получали следующее соединение 21(Е)-9-О-2 метил-6 метил(4-трифторметилфенил)метиламиногексиламино этил оксим эритромицина А (соединение 39), МС (МН)1078 13 С-ЯМР (50 МГц,3)(част./млн) 143,65 129,12 129,03 125,10 124,29. Пример 23. Фармакологическая активность а) Антибактериальная активностьОпределение минимальных ингибирующих концентраций (МИК) по отношению к грамположительным и грамотрицательным бактериям осуществляли микробиологическим методом серийных двукратных постепенных разведений жидкой среды, 19907-2-, ,с использованием в качестве среды для культивирования бульона Мюллера-Хинтона(МНВ). В случае требовательных бактерий (и) в среду добавляли 5 -ную лошадиную сыворотку. В качестве эталонных соединений из класса макролидов применяли рокситромицин и кларитромицин,изд.,8253 и 2340 соответственно. МИК, выраженные в (мкг/мл), определяли после инкубации микропланшетов при 37 С в течение 18 часов, оценивая наименьшую концентрацию антибиотика, обладающую способностью ингибировать развитие бактерий. б) Антибактериальная активностьТерапевтическую эффективность, выраженную в виде средней защищающей дозы(50) исследуемых соединений формулы , оценивали на примере индуцированной экспериментальным путем легочной инфекции, вызванной у мышиС 203. Использовали мышей-альбиносов линии(штамм 1) с весом тела 2335 г, которых помещали в клетку группами по 6 особей, давая стандартный корм и воду по желанию. Суспензию .С 203 (соответствующую приблизительно 108 ) в триптозном бульоне (0,05 мл) вводили интраназально каждой мыши, анестезированной смесью этилового эфира и хлороформа. Исследуемые соединения вводили внутрибрюшинно в виде однократной дозы в 0,2 -ной суспензии Твина за 24 часа до или через 1 час после заражения. Наблюдение за гибелью мышей осуществляли в течение периода времени до 10 дней после заражения. Определение 50, выраженной в виде (мг/кг), проводили методом пробит-анализа. Данные об антибактериальной активности в опытахпо отношению к грамположительным микроорганизмам (таблица 1) и грамотрицательным микроорганизмам (таблица 2) и об антибактериальной активности в опытах(таблица 3) некоторых характерных соединений формулыприведены ниже. Приведенные в таблице 1 данные явно свидетельствуют о том, что соединения формулы , являющиеся предметом настоящего изобретения, обладают антибактериальной активностью по отношению к грамположительным микроорганизмам, практически сопоставимой с таковой рокситромицина и кларитромицина. Антибактериальная активность соединений формулыпо отношению к грамотрицательным микроорганизмам, таким каки, оказалась существенно выше, чем таковая обоих эталонных соединений. Соединения формулыоказались активными в опытах, а профили их активности свидетельствуют о том, что эти соединения обладают существенно более высокими продолжительностью действия и периодом полураспада при элиминации из ткани, чем таковые соединений-эталонов. 4962 1 Таблица 1 Антибактериальная активность соединений 2, 4-12, 16-19, 21, 23-28 и соединенийэталонов рокситромицина и кларитромицина в опытахотношению к грамположительным микроорганизмам, таким как 3,4,26,С 203,29212 и 14, выраженная в виде минимальной ингибирующей концентрации МИК (мкг/мл) МИК (мкг/мл) 4962 1 Таблица 2 Антибактериальная активность соединений 2, 4-12, 16-19, 21, 23-38 и соединенийэталонов рокситромицина и кларитромицина в опытахп отношению к грамотрицательным микроорганизмам, таким какАТСС 25922 и 2, выраженная в виде минимальной ингибирующей концентрации МИК (мкг/мл). Соединение 2 4 5 6 7 8 9 10 11 12 16 17 18 19 21 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 Рокситромицин Кларитромицин 4962 1 Таблица 3 Антибактериальная активность в опытахсоединений 4, 10, 16-19, 21, 23, 26-27,29-30, 33-35, 38 и соединений-эталонов рокситромицина и кларитромицина при введении за 24 часа до и через 1 час после заражения мыши экспериментальным путем легочной инфекцией, вызваннойС 203, выраженная в виде средней защищающей дозы 50 (мг/кг). Соединение 4 10 16 17 18 19 21 23 26 27 29 30 33 34 35 38 Рокситромицин Кларитромицин Национальный центр интеллектуальной собственности. 220034, г. Минск, ул. Козлова, 20.
МПК / Метки
МПК: C07H 17/08, A61P 31/04, A61K 31/7048
Метки: эритромицина, 9-o-оксимовые, композиция, фармацевтическая, производные
Код ссылки
<a href="https://by.patents.su/25-4962-9-o-oksimovye-proizvodnye-eritromicina-a-i-farmacevticheskaya-kompoziciya.html" rel="bookmark" title="База патентов Беларуси">9-O-Оксимовые производные эритромицина А и фармацевтическая композиция</a>
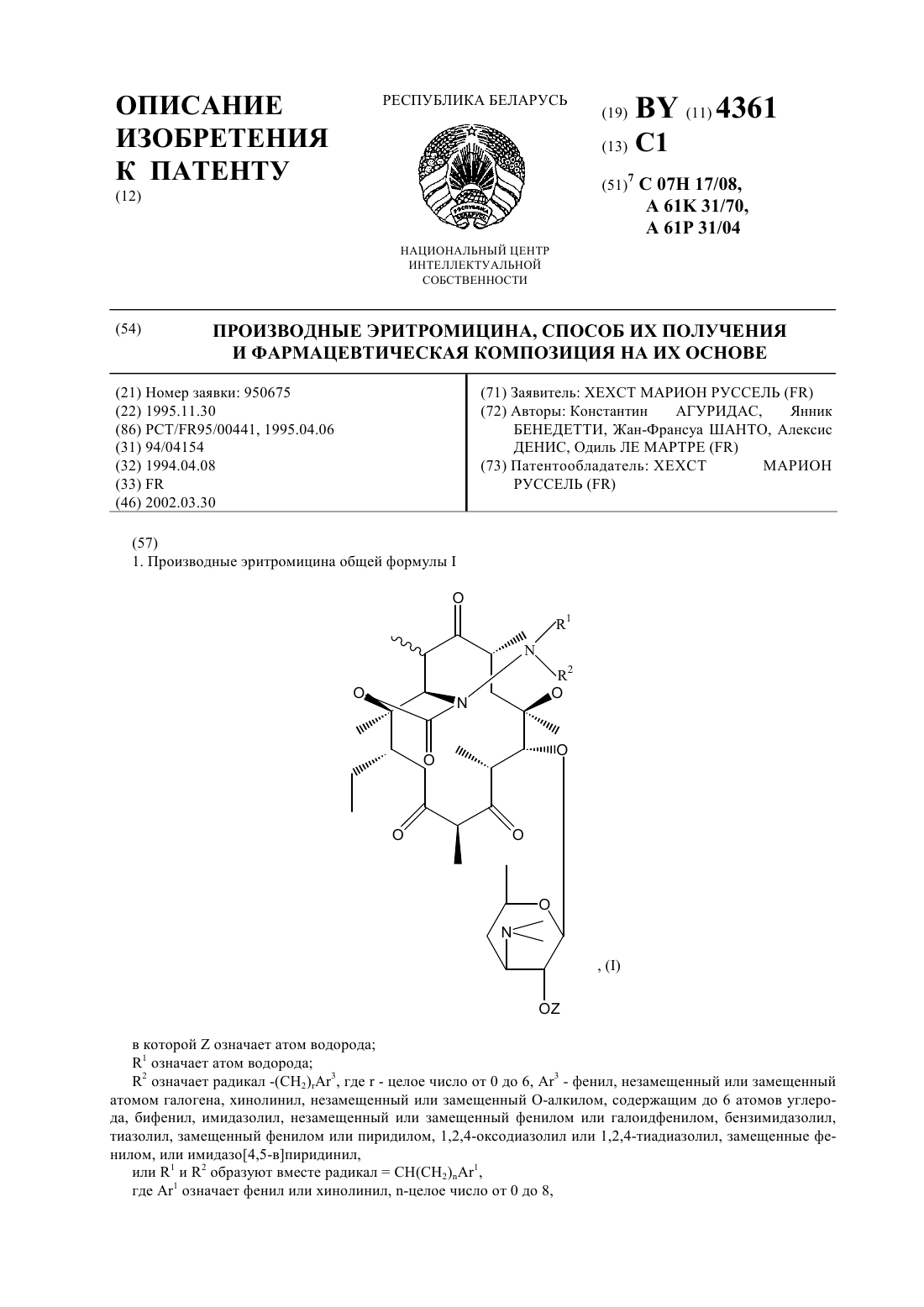
Производные эритромицина, способ их получения и фармацевтическая композиция на их основе
Номер патента: 4361
Опубликовано: 30.03.2002
Авторы: Константин АГУРИДАС, Одиль ЛЕ МАРТРЕ, Янник БЕНЕДЕТТИ, Алексис ДЕНИС, Жан-Франсуа ШАНТО
МПК: C07H 17/08, A61K 31/70, A61P 31/04...
Метки: эритромицина, основе, получения, производные, фармацевтическая, композиция, способ
Текст:
...соли калия, которую растворяют в 1,2 л воды, подкисляют до рН 1 раствором концентрированной соляной кислоты после фильтрования, получают 29 г целевого продукта рассчитанный 58,52 3,43 6,82 15,6 обнаруженный 58,5 3,7 6,8 15,2. Стадия С 2-фенил-5-тиазолкарбоксилатметил. К раствору 4,77 г кислоты, полученной на стадии В, в 160 мл метанола, прибавляют 2,5 мл ацетилхлорида и осуществляют нагрев при рефлюксе в течение 18 ч. При пониженном...
Производные эритромицина, способ их получения, фармацевтическая композиция на их основе и промежуточные соединения
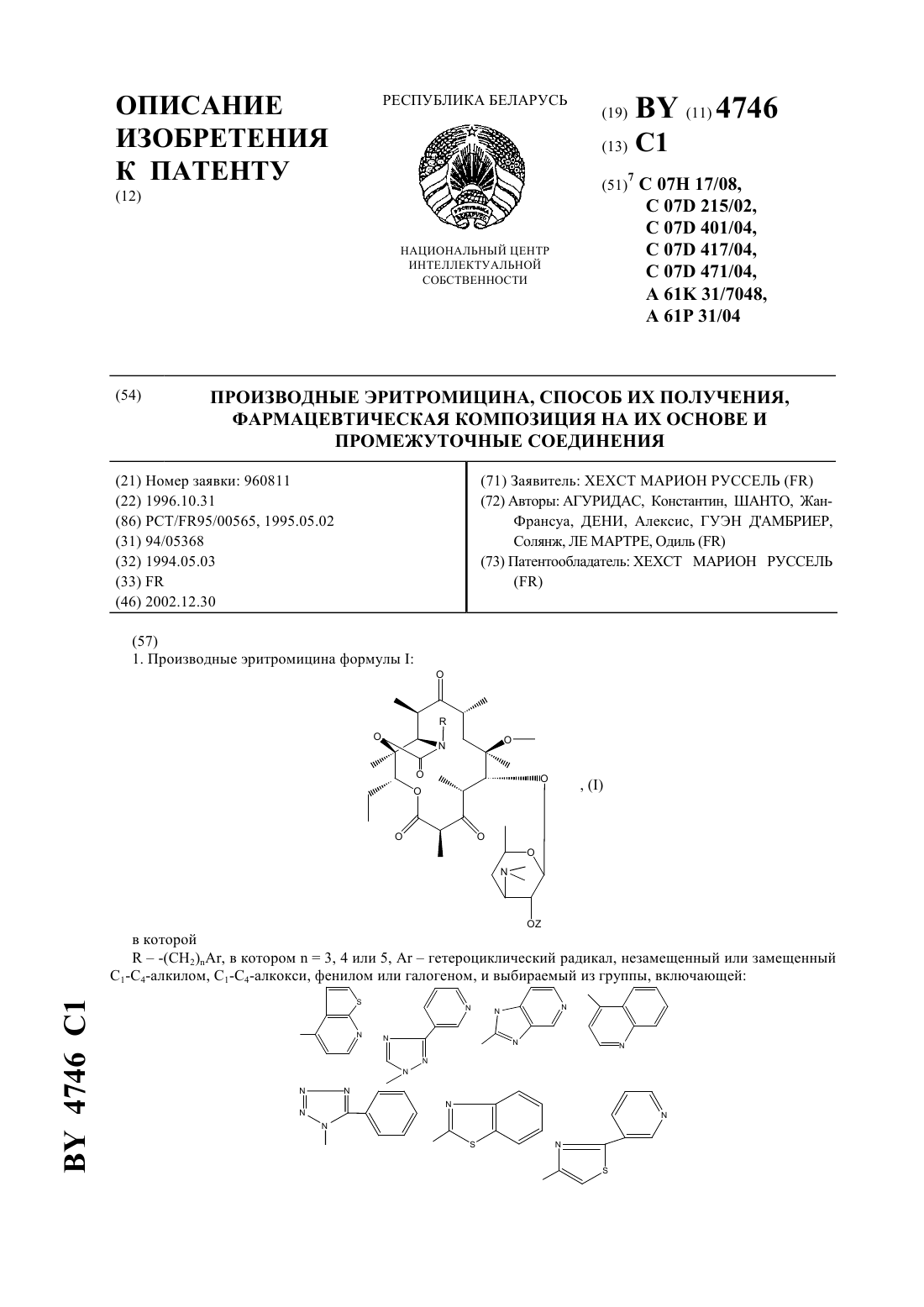
Номер патента: 4746
Опубликовано: 30.12.2002
Авторы: ГУЭН Д'АМБРИЕР, Солянж, ШАНТО, Жан-Франсуа, АГУРИДАС, Константин, ДЕНИ, Алексис, ЛЕ МАРТРЕ, Одиль
МПК: A61K 31/7048, C07D 215/02, A61P 31/04...
Метки: композиция, получения, производные, эритромицина, фармацевтическая, промежуточные, способ, основе, соединения
Текст:
...2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(2-фенил-4-хинолинил)бутил)иминоэритромицин. Т. пл.195-197 . Пример 20. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6 метил-3-оксо 12,11-(оксикарбонил 4-(1-бензотриазол-1-ил)бутил)иминоэритромицин. Т. пл.200-202 . Пример 21. 11,12-дидеокси-3-де 2,6-дидеокси-3 метил-3 метил-альфарибогексопиранозил)окси)-6...
Производные пенема, способы их получения и фармацевтическая композиция на их основе

Номер патента: 4096
Опубликовано: 30.09.2001
Авторы: Федерико Мария АРКАМОНЕ, Пьеро СБРАЧИ, Витторио ПЕСТЕЛЛИНИ, Мария АЛЬТАМУРА, Энцо ПЕРРОТТА, Джузеппе КАСЧЬО
МПК: A61P 31/04, C07D 499/883, A61K 31/43...
Метки: производные, способы, основе, композиция, получения, пенема, фармацевтическая
Текст:
...активный агент и фармацевтически приемлемый носитель, отличающаяся тем, что в качестве активного агента включает соединение по п. 1 в эффективном количестве.9. Фармацевтическая композиция по п. 8, отличающаяся тем, что дополнительно включает антибиотики или ингибиторы бета-лактама.Настоящее изобретение относится к производным пенема общей формулы 1112 - карбоксильная группа или этерифицированная карбоксильная группа, легко активируемая...
Производные 2-амино-1,2,3,4-тетрагидронафталина и фармацевтическая композиция, обладающая кардиоваскулярной активностью

Номер патента: 3782
Опубликовано: 30.03.2001
Авторы: КАСАГРАНДЕ, Чезаре, СЕМЕРАРО, Клаудио, МОНТАНАРИ, Стефания, МАРЧИНИ, Франческо, САНТАНЖЕЛО, Франческо, БЕРТОЛИНИ, Джорджио
МПК: A61P 9/04, A61P 9/12, A61K 31/135...
Метки: композиция, кардиоваскулярной, активностью, обладающая, производные, 2-амино-1,2,3,4-тетрагидронафталина, фармацевтическая
Текст:
...хлористым метиленом. Полученную органическую фазу сушат над безводным сульфатом натрия и растворитель испаряют. Очисткой полученного сырого продукта колоночной хроматографией на силикагеле (230-400 меш) с элюированием смесью хлористый метилен-метанол (91) получают заглавное соединение (20 г) с т. пл. 69-70 С (этилацетат). 7 Н-ЯМР (300 МГц, 3)(ч/млн)1,37 (м, 2 Н), 1,5-1,7 (м, 4 Н), 2,33 (т, 2 Н), 3,33 (м, 2 Н), 3,88 (с, 3 Н),4,55 (с,...
Производные пирролзамещенных уреидов, способ их получения и фармацевтическая композиция на их основе

Номер патента: 2226
Опубликовано: 30.09.1998
Авторы: Марина ЧЬОМЕИ, Мариа Гранди, Никола МОНДЖЕЛЛИ, Джованни Бьясоли, Альфредо Пайо
МПК: A61K 31/40, C07D 207/34, C07D 207/335...
Метки: уреидов, основе, пирролзамещенных, способ, производные, фармацевтическая, получения, композиция
Текст:
...этилолеат, гликоли, такие как пропиленгликоль, и по желанию, соответствующее количество гидрохлорида лидокаина. Лекарственные средства для наружного применения могут быть изготовлены в виде кремов, примочек или паст для кожной обработки, например путем смешивания активного ингредиента со стандартными масляными или эмульгирующими наполнителями. Лекарственные средства для перорального введения могут быть изготовлены в виде таблеток и...
Предыдущий патент: Способ и соединения для магниторелаксометрического обнаружения аналитов и их применение
Следующий патент: Способ получения сапропелевого порошка для буровых растворов
Случайный патент: Способ сжигания топлива