Аналог инсулина
Номер патента: 3225
Опубликовано: 30.03.2000
Авторы: РОНАЛЬД ЮДЖИН ЧАНС, ДЖЕЙМС ЭДВИН ШИЛДЗ, РИЧАРД ДЕННИС ДИМАРЧИ, БРЮС ХИЛЛ ФРЭНК




























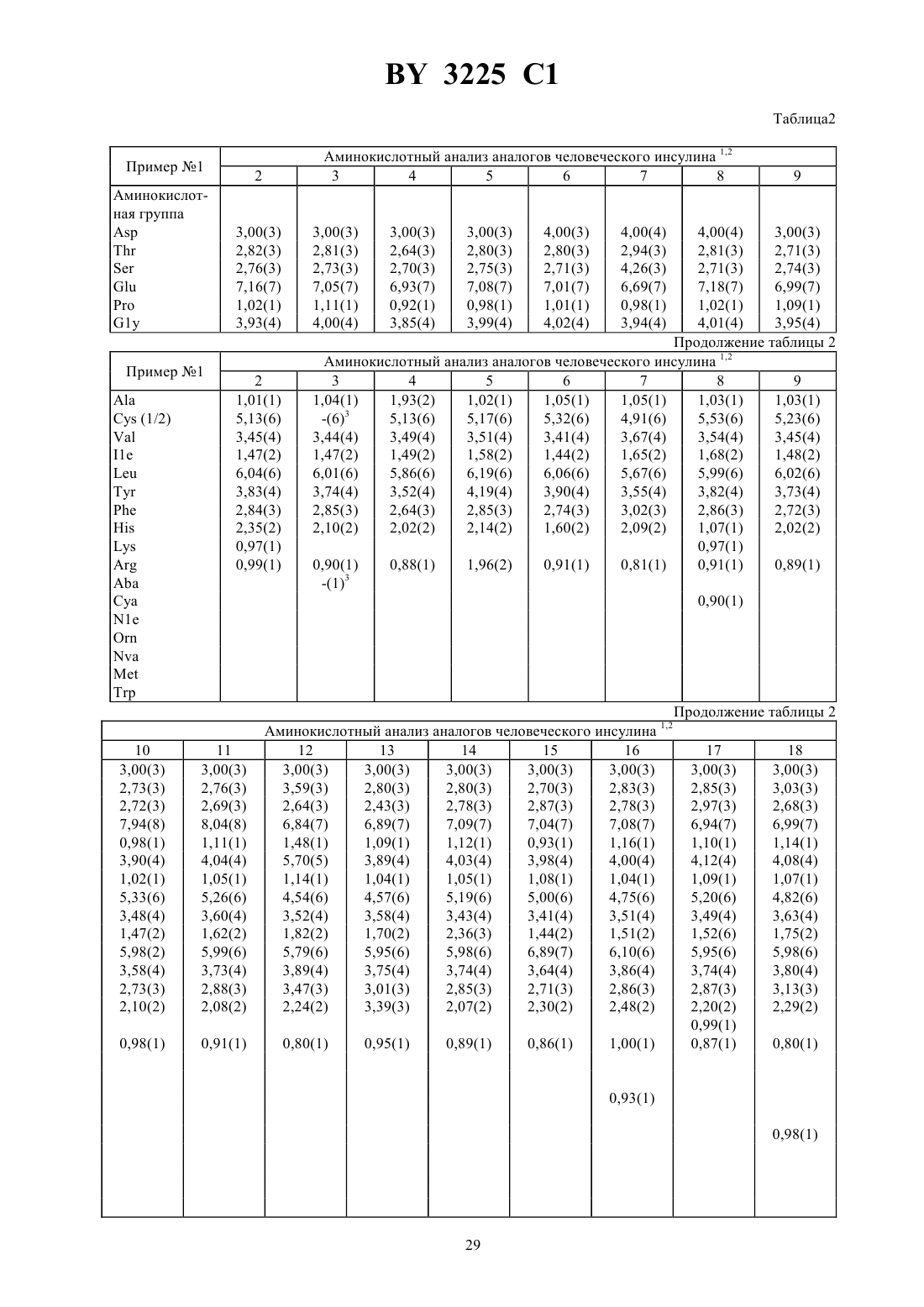
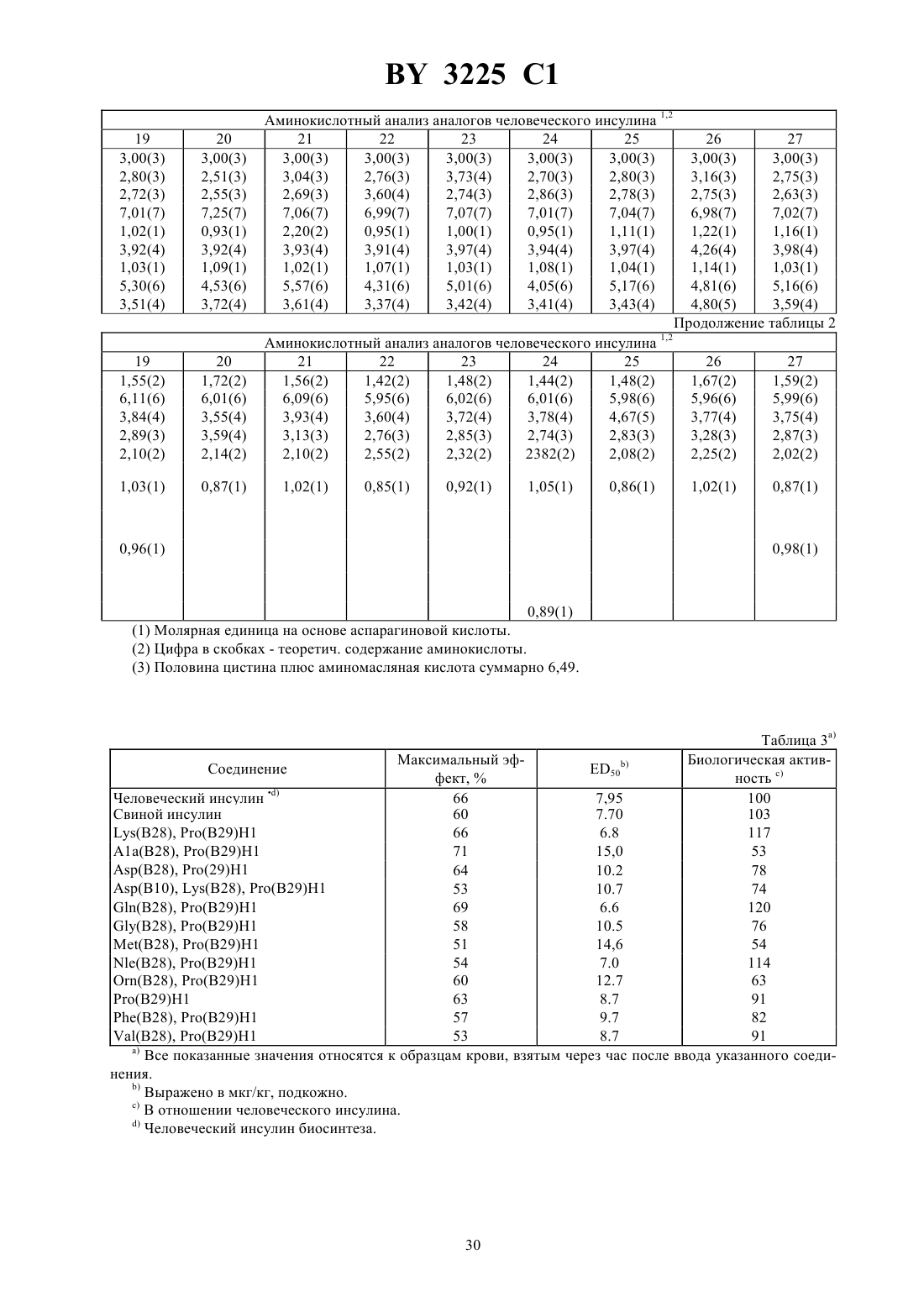
Текст
ГОСУДАРСТВЕННЫЙ ПАТЕНТНЫЙ КОМИТЕТ РЕСПУБЛИКИ БЕЛАРУСЬ(71) Заявитель ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ(72) Авторы Рональд Юджин Чанс, Ричард Деннис Димарчи, Брюс Хилл Фрэнк, Джеймс Эдвин Шилдз(73) Патентообладатель ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ(57) 1. Аналог инсулина формулы Фиг. 1 или его фармацевтически приемлемая соль, отличающийся тем, что А 21 является аспарагином В 1 является фенилаланином В 2 является валином В 3 является аспарагином В 9 является серином В 10 является гистидином или аспарагиновой кислотой В 27 является треонином В 28 является любой природной -аминокислотой В 29 является -пролином В 30 является треонином. 2. Аналог инсулина по п. 1, отличающийся тем, что В 28 является аспарагиновой кислотой, валином, лейцином, изолейцином, норлейцином, пролином, аргинином, гистидином, цитруллином, орнитином, лизином,фенилаланином, аланином или глицином. 3. Аналог инсулина по п. 1 или 2, отличающийся тем, что В 28 является аспарагиновой кислотой, валином, лейцином, изолейцином, норлейцином, пролином, аргинином, гистидином, орнитином или лизином. 4. Аналог инсулина по любому из пп. 1-3, отличающийся тем, что В 28 является лизином. 3225 1 5. Аналог инсулина по любому из пп. 1-4, отличающийся тем, что В 10 является аспарагиновой кислотой. 6. Аналог инсулина по п. 1, отличающийся тем, что А 21 является аспарагином В 1 является фенилаланином В 2 является валином В 3 является аспарагином В 9 является серином В 10 является гистидином В 27 треонином В 28 - лизином В 29 - -пролином В 30 - треонином. 7. Аналог инсулина формулыпо п. 1, обладающий активностью снижения уровня глюкозы в крови. Изобретение касается аналогов инсулина, модифицированного в положении 29 аминокислоты В-цепи естественного человеческого инсулина и при желании в других положениях. Эти аналоги инсулина в меньшей степени склонны к димеризации или самопроизвольной ассоциации с более высокомолекулярными формами, в результате чего они быстрее повышают свою активность, сохраняя при этом биологическую активность естественного человеческого инсулина. Изобретение охватывает аналоги инсулина формулы , приведенной на фиг. 1, в которой А 21 представляет собой аланин, аспарагин, аспарагиновую кислоту, глутамин, глутаминовую кислоту, глицин, треонин или серин В 1 представляет собой фенилаланин В 2 представляет собой валин В 3 представляет собой аспарагин В 9 представляет собой серин В 10 представляет собой гистидин или аспарагиновую кислоту В 27 представляет собой треонин В 28 представляет собой любую -аминокислоту В 29 представляет собой пролин. Описан также способ лечения гипергликемии путем ввода в организм пациента, нуждающегося в таком лечении, эффективного количества аналога инсулина формулы . Кроме того, описаны фармацевтические композиции, содержащие эффективное количество аналога формулыв комбинации с одним или несколькими фармацевтически пригодными эксципиентами. На фиг. 2 изображены ограничительная точка и функциональная карта плазмиды рКС 283 на фиг. 3 - ограничительная точка и функциональная карта плазмиды рКС 283 РХ на фиг. 4 - ограничительная точка и функциональная карта плазмиды 283- на фиг. 5 - ограничительная точка и функциональная карта плазмиды рКС 283- на фиг. 6 - ограничительная точка и функциональная карта плазмиды рКС 283- на фиг. 7 - ограничительная точка и функциональная карта плазмиды 32 на фиг. 8 - ограничительная точка и функциональная карта плазмиды 789 на фиг. 9 схематически представлено построение плазмиды 120 на фиг. 10 изображены ограничительная точка и функциональная карта плазмиды 47 на фиг. 11 - ограничительная точка и функциональная карта плазмида 12 на фиг. 12 - ограничительная точка и функциональная карта плазмиды 121 на фиг. 13 - ограничительная точка и функциональная карта плазмиды 110 на фиг. 14 и 15 схематически представлено построение плазмиды 110 на фиг. 16 изображены ограничительная точка и функциональная карта плазмиды 126 на фиг. 17 показана нуклеотидная последовательность гена синтезированного человеческого проинсулина на фиг. 18 изображены ограничительная точка и функциональная карта плазмиды рРВ 145 на фиг. 19 - ограничительная точка и функциональная карта плазмиды 164 на фиг. 20 - ограничительная точка и функциональная карта плазмиды 172 на фиг. 21 - ограничительная точка и функциональная карта плазмиды 173 на фиг. 22 - ограничительная точка и функциональная карта плазмиды 175. Формулавоспроизводит с помощью сокращенных названий аминокислотную последовательность аналогов инсулина, отвечающих данному изобретению. Сокращенные названия аминокислот могут быть расшифрованы, как указано ниже. Аббревиатура Аминокислота Треонин Тр Триптофан Ту Тирозин Валин В 28 может представлять собой любую аминокислоту природного или неприродного происхождения. Эта аминокислота является предпочтительно аспарагиновой кислотой, валином, лейцином, изолейцином, норлейцином,пролином, аргинином, гистидином, цитруллином, орнитином, лизином, фенилаланином, аланином или глицином. Из указанных выше аминокислот особенно предпочтительной подгруппой является аспарагиновая кислота, валин,лейцин, изолейцин, норлейцин, пролин, аргинин, гистадин, орнитин или лизин. Лизин является самой предпочтительной аминокислотой для В 28. В 29 представляет собой предпочтительно -пролин. Особенно предпочтительным аналогом инсулина, согласно изобретению, является такой, в котором В 28 представляет собой лизин и В 29 представляет собой пролин, то есть инверсию аминокислотной последовательности естественного человеческого инсулина в положениях 28 и 29 В-цепи. Согласно следующей особенности изобретения, как указывалось выше, когда В 28 представляет собой пролин, то положение В 29 представляет собой предпочтительно -(-метиллизин), -лизин, -(-метиларгинин) или -аргинин. Предпочтительным аналогом, согласно данной особенности, является такой, в котором В 28 представляет собой -пролин и В 29 представляет собой -(-метиллизин) или -лизин. Предпочтительным значением концевой группы является НООС. Ниже рассматриваются также другие модификации аналогов инсулина согласно изобретению, то есть модификации аналогов инсулина в положениях иных, чем положения В 28 и В 29. В частности, может быть желаемой замена группыв положении 21 цепи А (то есть карбоксильной концевой группы) группами, , , , ,или , и при такой замене предпочтительной замещающей группой является. Аналогично этому может быть желательной замена группы в положении 3 цепи В аспарагиновой кислотой . Такие возможные замены приводят к увеличению стабильности аналогов при экстремальных значениях рН, посколькуособенно чувствителен к реакциям деамидирования и перегруппировки как при низком, так и при высоком значении рН. Для специалистов в данной области должно быть ясно также, что глутаминовые группы аналогов инсулина могут быть одинаковым образом чувствительны к реакциям деамидирования и перегруппировки. Возможные другие модификации аналогов инсулина включают (в любой комбинации) замену гистидиновой группы в положении В 10 аспарагиновой аминокислотой, замену фенилаланиновой группы в положении А 1 аспарагиновой кислотой, замену треониновой группы в положении В 30 аланином, замену сериновой группы в положении В 9 аспарагиновой кислотой, делецию аминокислот в положении В 1 (-1) либо как таковых, либо в комбинации с делецией в положении В 2 (-2) и делецию треонина из положения В-30 (-В 30). Аналоги инсулина могут быть также модифицированы в концевых положениях В 30 путем ввода любых из следующих аминокислот или дипептидов , -, , -, - или -. Предпочтительным из них является -. Кроме того, в случае наличия этих добавленных звеньев в положении В 30 аналог может быть дополнительно модифицирован путем добавления к образуемой В 30-удлиненной концевой группе глутаминовой кислотыили аминокислотной последовательности, включающей всю или часть последовательности , которая начинается в ее -терминальной -. Когда эта последовательность представляет лишь часть указанной выше, то она может представлять собой любую из тех частей, которые начинаются у -конца данной последовательности, то есть у группы глутаминовой кислоты . Комбинация аминокислот, которые определены выше, представляет собой полностью или часть соединяющего пептида, обнаруживаемого в человеческом проинсулине, молекуле, которая является биологическим источником образования естественного человеческого инсулина. Предпочтительными последовательностями являются 1 . Как уже упоминалось выше, изобретение охватывает фармацевтически пригодные соли аналогов инсулина. Такими предпочтительными солями являются соли цинка, натрия, калия, магния и кальция. Аналоги инсулина, отвечающие данному изобретению, могут быть получены одним из известных приемов синтеза пептидов, включая классические способы (в растворе), способы в твердофазных системах, способы полусинтеза и известные с недавнего времени способы рекомбинантной ДНК. В способах с использованием твердофазных систем аминокислотная последовательность составляется из исходной нерастворимой С-терминальной аминокислоты со смоляным носителем. Эти твердофазные способы описываются .и др. Твердофазный синтез пептидов.., Сан-Франциско, 1969 г. Обычно при осуществлении твердофазного способа аминокислота, соответствующая С-терминальной аминокислотной группе желаемого пептида, закрепляется на смоляном носителе и далее формируется пептидная цепь, начинающаяся с нанесенной на смолу С-терминальной аминокислоты. Отдельные аминокислоты вводятся последовательно до тех пор, пока не получается желаемая аминокислотная последовательность. Как возможный вариант могут быть получены небольшие пептидные фрагменты, которые могут быть введены в пептидную цепь в желаемом порядке. Пептидная цепь остается скрепленной со смолой в процессе синтеза, и по завершении построения цепи данный пептид отщепляется от смолы. Пептидная цепь закрепляется с полистирольной смолой посредством сложноэфирной связи, образующейся между карбоксильной группой С-терминального звена и специфической метиленовой группой, присутствующей в смоляной матрице, являющейся точкой такого скрепления. Аминокислоты связываются сопряженными связями хорошо известными в данной области приемами образования пептидных связей. Один такой прием заключается к конверсии аминокислоты в производное, которое придает карбоксильной группе более высокую чувствительность к реакции со свободной -терминальной аминогруппой пептидного фермента. Так, например, аминокислота может быть превращена в смешанный ангидрид путем реакции защищенной аминокислоты с этилхлорформатом, фенилхлорформатом, вторбутилхлорформатом или изобутилхлорформатом. Как возможный вариант аминокислота может быть превращена в активный сложный эфир, такой как 1,4,5-трихлорфениловый сложный эфир, пентахлорфениловый сложный эфир, паранитрофениловый сложный эфир, -оксисукцинимидный сложный эфир или сложный эфир, образуемый из 1-оксибензотриазола. Другой прием сопряжения заключает в себе использование подходящего агента, такого как , дихлоргексилкарбодиимид (ДСС) или , -диизопропилкарбодиимид . Другими подходящими сопрягающими агентами являются соединения, которые уже известны для специалистов в данной области (см. работуиПептиды,, 1965 г. Глава ). Следует иметь в виду, что -аминогруппа каждой аминокислоты, используемой в синтезе пептидов,должна быть защищена в ходе реакции сопряжения для предотвращения побочных реакций, в которых принимает участие -аминовая функциональная группа. Следует также иметь в виду, что некоторые аминокислоты содержат боковые цепи, являющиеся функциональными группами (например, сульфгидрил, -амино, и -карбоксил, имидазол, гуанидино и гидрокси), и что такие функциональные группы также должны быть защищены в начальном и в последующих этапах реакции сопряжения. Подходящие защитные группы известны в данной области (Защитные группы в органической химии. - . . Ред.. -4,1973, и патент США 4617149). При выборе типа защитной группы следует соблюдать определенные условия. Защитная для -амино группа должна придавать функциональной -аминогруппе инертность в условиях, имеющих место в реакции сопряжения, должна быть легко удаляемой после реакции сопряжения в таких условиях, в которых не удаляются защитные группы боковой цепи, не должна изменять структуру пептидного фрагмента и должна исключать возможность рацемизации при активации непосредственно перед реакцией сопряжения. Защитная группа боковой цепи должна придавать этой функциональной боковой цепи инертность в условиях, используемых при протекании реакции сопряжения, должна быть устойчивой в условиях, имеющих место при удалении защитной для -амино группы, и должна легко удаляться по завершении построения желаемой аминокислотной последовательности в таких условиях реакции, которые не изменяют структуру пептидной цепи. Для специалистов в данной области должно быть ясно, что защитные группы, которые уже известны как группы, применяемые для синтеза пептидов, будут по-разному активны к агентам, используемым для их удаления. Так, например, некоторые защитные группы, такие как трифенилметил и 2-(пара-бифенил) изопропилоксикарбонил, очень неустойчивы и могут расщепляться в слабо кислотных условиях. Другие защитные группы, такие как трет-бутилоксикарбонил, трет-аминоксикарбонил, адаматнилоксикарбонил и параметоксибензилоксикарбонил, менее устойчивы и требуют использования умеренно сильных кислот, таких 4 3225 1 как трифторуксусная кислота, соляная кислота или трифтор бора в уксусной кислоте, для их удаления. Некоторые защитные группы, такие как бензилоксикарбонил, галоилбензилоксикарбонил, пара-нитробензилоксикарбонил, циклоалкилоксикарбонил и изопропилоксикарбонил, еще менее устойчивы и для их удаления требуется использование еще более сильных кислот, таких как фтористоводородная кислота, бромистоводородная кислота или трифторацетат бора в трифторуксусной кислоте. По завершении желаемого построения последовательности пептида защищенный пептид должен быть отщиплен от смоляного носителя и все защищающие группы должны быть удалены. Реакция отщепления и удаление защитных групп могут осуществляться одновременно или поэтапно. Когда смоляной носитель представляет собой хлорметилированную полистироловую смолу, то связь, скрепляющая пептид со смолой,является сложноэфирной связью, образуемой между свободной карбоксильной группой С-терминального звена и одной из многих хлорметильных групп, присутствующих в смоляной матрице. Следует иметь в виду, что указанная скрепляющая связь может быть расщеплена посредством реагентов,которые, как известно, способны разрывать сложноэфирную связь и проникать в смоляную матрицу. Особенно легко осуществимым способом является обработка жидким безводным фтористым водородом. Этот реагент не только отщепляет пептид от смолы, но и удаляет все защитные группы. Следовательно, использование этого реагента может обеспечить получение полностью лишенного защиты пептида. Когда желательно удаление пептида без удаления защитных групп, то защищенный пептид - смола может подвергаться метанолизу, в результате чего получается защищенный пептид, в котором С-терминальная карбоксильная группа подвергнута метилированию. Этот сложный метиловый эфир затем может быть гидролизован в слабо щелочных условиях, и в результате будет получен свободный С-терминальный карбоксил. Защитные группы пептидной цепи могут быть удалены путем обработки сильной кислотой, такой как жидкая фтористоводородная кислота. Особенно интересный способ метанолиза описывается в работе .и др. Пептиды. Труды 5-го Американского симпозиума по пептидам. - .и . , ред.. -9.,1977. - С. 518 - 521, где замещенный пептид-смола обрабатывается метанолом и цианидом калия в присутствии простого кроун-эфира. Следующий способ отщепления защищенного пептида от смолы заключается в аминолизе или обработке гидразином. При желании образуемый С-терминальный амид или гидразид может быть гидролизован до свободного С-терминального карбоксила, и защитные группы могут быть удалены общепринятым образом. Следует также иметь в виду, что защитные группы, находящиеся у -терминальной -аминогруппы, удаляются предпочтительно либо до, либо одновременно с отщеплением защищенного пептида от смоляного носителя. А- и В-цепи аналогов инсулина, отвечающие данному изобретению, также могут быть получены методом рекомбинантной ДНК. При этом получается нуклеотидная последовательность, кодирующая желаемый пептид А- или В-цепи, с использованием общепринятой технологии для такого синтеза. Эти методы обычно заключают в себе получение олигонуклеотидов, кодирующих как фрагменты желаемой кодирующей последовательности, так и их полную последовательность. Олигонуклеотиды обеспечивают перекрытие одного фрагмента кодирующей последовательности двумя фрагментами дополняющей последовательности и наоборот. Эти олигонуклеотиды конъюгируют и связываются с образованием в конечном итоге желаемой генной последовательности. Эта последовательность вставляется в клонирующий вектор в точке, которая допускает возможность выражения пептидного продукта, который он кодирует. Подходящий клонирующий вектор заключает в себе, по меньшей мере, часть контрольной последовательности выражения гена. Цепи А и В аналогов инсулина, согласно изобретению, могут быть также получены посредством исходной молекулы проинсулинового типа с использованием методов рекомбинантной ДНК (см.и др. Пептиды Синтез - Структура - функции. Труды 7-го Американского симпозиума по пептидам. Ред. .и .(1981 г.). Однако этап комбинирования отдельных получаемых цепей А и В может осуществляться способом, описанным в работеи др., Пептиды Синтез, структура и функции. Труды 7-го Американского симпозиума по пептидам, 1981 г. Для иллюстрации данного изобретения даются нижеследующие примеры, которые, однако, не служат для ограничения объема изобретения. Пример 1. Человеческий инсулин(В 29),(В 28). А. Получение образованной от рекомбинанты А-цепи. А-цепь человеческого инсулина получается методом рекомбинантной ДНК путем химического синтеза гена, кодирующего А-цепь, и его выражения в . . Ген для А-цепи синтезируется из различных тринуклеотидов, синтетические фрагменты, составляющие по длине от дека до пентадекануклеотидов, посредством метода получения блок-фосфотриэфира. Этот ген имеет однониточный когезионный конец для ограничительных эндонуклеази Ва . Вставка его в подходящий выраженный вектор, содержащий галактозный ген (-), приводит к химерной плазмиде, содержащей А-цепь, связанную с геном - через метиониновый кодон. Для достижения более высоких уровней выражения используется предпочтительно ген 5 3225 1 триптофан синтетазыв качестве промотора, а не ген. Химерная плазмида трансформируется в. , приводящей в результате к выражению исходного белка (белка предшественника), то есть -А-цепи (или- А-цепи, когда используется система промотора). Обработка исходного белка цианогенбромидом приводит к расщеплению метиониновой связи с получением после очистки А-цепи человеческого инсулина. Окислительный сульфитолиз приводит к образованию -сульфонированной А-пепи, которая используется для комбинации с В-цепью (-сульфонат), как описано ниже. Подробное описание химического синтеза генов для А-человеческого инсулина дается в публикациии др. Рос. ., . ., США, 75, 57655769, 1978. Для получения полных сведений о выражении в .химически синтезированного гена для А-цепи человеческого инсулина см. публикациюи др. Рос.. . ., США, 76, 101-110, 1979. В. Получение аналога В-цепи(В 28),(В 29). Для получения сырой пептидиловой смолы используется синтезатор пептида 430 А(включая модификацию А 4). Используется 0,5 ммоль исходной твердофазной смолы ( (1) ОСН 2 Ра смолы) (0,76 ммоль/г 0,658 г). Все аминокислоты являются защищенными ВОС, и, кроме глутаминовой кислоты и гистидина, все аминокислоты используются непосредственно в том виде, как они получены(то есть в упаковках, содержащих примерно от 2 ммоль защищенной аминокислоты, поставляемых фирмой). Глутаминовая кислота и гистидин поставляются фирмой, и они заключаются в такие упаковки, что каждая содержит примерно 2 ммоль желаемой защищенной аминокислоты. После сушки сырой пептидиловой смолы (в вакууме при комнатной температуре в течение ночи) определяется ее вес и сопоставляется с начальным весом для гарантии нужного привеса. Небольшая часть образца подвергается аминокислотному анализу для гарантии того, что желаемые аминокислоты вводятся в точно заданных количествах. Пептид отщепляется от пептидиловой смолы, и защита с боковой цепи удаляется в результате перемешивания примерно в течение 1 ч при 0 С в растворе 10 частей (об./вес.)(содержащего 5 об./об. этилмеркаптана и 5 об./об. метакрезола) с 1 частью пептидиловой смолы. После вакуумной отгонки большей частипептид осаждается в простом этиловом эфире. После нескольких промывок простым этиловым эфиром с последующей вакуумной фильтрацией пептид растворяется примерно в 200 мл 7 М деионизированной воды, содержащей 0,1 моль Трис, 0,1 моль 23 и 0,01 моль 24 О 6. Величина рН раствора доводится до 8,5 посредством 5 н. , и этот раствор интенсивно перемешивается в течение ночи при температуре 4 С. Образующийся -сульфонированный пептидный раствор вводится в колонку размерами 5215 см, наполненную-25 (тонкоизмельченным) при комнатной температуре. Образец элюируется 50 мМ бикарбонатом аммония с расходом его 20 мл/мин при комнатной температуре. Выходящий поток анализируется (при 276 нм). Фракции по 20 мл собираются, и получают объем желаемых фракций, который затем очищается дополнительно методом жидкостной хроматографии высокого разрешения , как описано ниже. Слитые желаемые фракции нагнетаются в колонку 2,530 см сС-8, 9-12 мкм (колонка ), и осуществляется элюирование с использованием линейного ингредиента с увеличивающейся концентрацией ацетонитрила в 100 мл бикарбоната аммония при комнатной температуре (2,6 мл/мин). Выходящий из колонки поток анализируется (280 нм). Фракции по 25 мл собираются. Выбранные фракцииподвергаются анализу для определения того, какие фракции остаются. Желаемые фракции собираются, лиофилизируются и используются в последующей комбинации с А-цепью, полученной, как описано выше. С. Получение человеческого инсулина(В 29),(В 28). Комбинация цепи А и В осуществляется согласно процедуре, описанной в публикациии др., см. выше. 700 мг -сульфоната А-цепи, образованного от рекомбинантной ДНК, и 140 мг синтетического сульфоната В-цепи(В 28),(В 29) (оба получены, как описано выше) каждый растворяются в 70 мл и 14 мл соответственно 0,1 М буферного раствора глицина и величина рН каждого доводится до 10,5 посредством 4 н. , затем они охлаждают до 5 С. Приготавливается 6 мл раствора дитиотреитола (ДТТ) концентрацией 10,5 мг/мл в 0,1 М глициновом буферном растворе при комнатной температуре, величина рН этого раствора доводится до 10,5 посредством 5 н. , затем этот раствор охлаждается до 5 С. Эти растворы А-цепи и В-цепи смешиваются друг с другом, после чего в эту смесь быстро вводится 5,21 мл раствора ДТТ (/30,90). Этот реакционный раствор перемешивается при температуре 5 С в открытых склянках 200-миллилитровых пробирках для центрифуги в течение 2,5 ч при 5 С. Вводится 45 мл ледяной уксусной кислоты, и раствор выстаивается при температуре 5 С в течение ночи. Полученная смесь с осадком центрифугируется в течение 20 мин со скоростью 2000 об/мин при температуре 5 С. Поверхностный слой смешивается с 1 М уксусной кислотой, промывается от твердых частиц, вводится в колонку 5200 см с-50 (сверхтонкого) в 1 М уксусной кислоте при 5 С и элюируется при ускорении силы тяжести. Двадцатиминутные фракции элюирования собираются в течение 3 дн. Эти фракции анализируются (при 276 нм), и некоторые образцы анализируются методами аналитической . Фракции, содержащие последовательность(В 28),(В 29) аналога инсулина, сливаются и лиофилизи 6 3225 1 руются, давая 125-миллиграммовый образец. Этот образец дополнительно очищается методом жидкостной хроматографии высокого разрешенияс обратимой фазой (с использованием колонки 2,1225 см сС-8 при элюировании при комнатной температуре со скоростью 2,6 мл/мин с использованием линейного градиента с увеличивающейся концентрацией ацетонитрила в 0,1 М 24, рН 2,2). Выходящий поток анализируется (при 276 нм). Выбранные фракции анализируются аналитическим методом . Желаемые фракции собираются и дополнительно очищаются с использованием рН 7 , как описано ниже. Слитный продукт отнизкого рН разбавляется примерно в два раза в ледяной бане 0,1 М(4)2 О 4. Величина рН доводится до 7 посредством холодной 3 н.в ледяной бане. Этот образец вводится в ту же колонку и элюируется из нее в тех же условиях, что и при приготовлении образца с низким рН, с той разницей, что элюирующий буферный раствор в данном случае представляет собой 0,1 М (рН 7)(4)2 О 4/ацетонитрил. Слитый продукт отс рН 7 быстро охлаждается в ледяной бане и в два раза разбавляется 0,1 -ным водным раствором трифторуксусной кислоты . Вводится 1 н. НС 1 (холодная, образец в ледяной бане) для снижения величины рН до 3. Образец вводится в колонкуС 4 или, как возможный вариант, в колонку с-8(2,1225 см), и осуществляется элюирование линейным градиентом с повышающейся концентрацией ацетонитрила в 0,1 -ном водном растворе . Выходящий из колонки продукт анализируется (при 214 или 276 нм). Желаемые фракции сливаются и лиофилизируются, давая образец в количестве 41 мг желаемого аналога со степенью чистоты более 97 , достигаемой путемс обратимой фазой. Пример 2. Человеческий инсулин(В 29),(В 28). Второй способ получения человеческого инсулина(В 29),(В 28) заключается в использовании ферментного полусинтеза (обратимый протеолиз) для комбинирования - октапептидного инсулина (1-21 В 1-22) с синтетическим октапептидом. -Октапептидный инсулин получается в результате триптического переваривания естественного свиного или человеческого инсулина, как описано в работеи се Получение и свойства -октапептид-инсулина, .133, 219-223, (1967 г). Синтетический октапептид получается методом автоматического твердофазного синтеза так, как описано выше.-октапептидный инсулин (435 мг) и синтетический октапептид (465 мг) смешиваются в 15 мл раствора, содержащего 1 часть диметил-сульфоксида, 2 части 1,4-бутандиола и 1 часть 0,25 М буферного раствора Трис-ацетата, рН 7,3. Пептиды полностью растворяются путем нагревания раствора на горячей плите. Затем раствор инкубируется при 37 С и добавляется 90 мг свиного трипсина. Раствор время от времени перемешивается в течение 90 мин при температуре 37 С. Реакция прекращается путем смешивания раствора с 135 мл 0,05 н. НС 1. Указанный в названии аналог инсулина очищается путем ввода подкисленного раствора, содержащего этот аналог, в колоннуразмерами 2,525 см, наполненную С-8 , и элюирования линейным градиентом с повышающейся концентрацией ацетонитрила в буферном растворе 0,1 М одноосновного фосфата натрия, рН 2,2. Выходящий на колонки продукт анализируется (при 276 нм). Желаемые фракции сливаются,в два раза разбавляются водой и вводится в колонкуразмерами 125 см с С-8 . Этот аналог элюируется линейным градиентом с увеличивающейся концентрацией ацетонитрила в 0,5 -ном водном растворе . Выходящий из колонки продукт анализируется (при 276 нм). Желаемые фракции снова сливаются и лиофилизируются, давая 125 мг очищенного аналога. Данная структура подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией .5809,2 (теоретически 5808,7). Осуществляется анализ методом быстрой атомной бомбардировки - масс-спектроскопии с использованием масс-спектрометра с двойной фокусировкой 25, с разрешением примерно 1500. Аналоги человеческого инсулина растворяются в смеси глицерина и триглицерина с содержанием щавелевой кислоты. Для калибровки измерительного прибора, который осуществляет магнитное сканирование в диапазоне от/ 5300 до / 6500, используется иодид цезия. Полученные данные представлены как среднемассовое значение 1. Пример 3. Человеческий инсулин(В 29),(В 28). Свиной -октапептидный инсулин (384 мг) и синтетический октапептид (362 мг) смешиваются в 13 мл раствора, содержащего диметилсульфоксид, 2 части 1,4-бутандиола и 1 часть буферного раствора 0,25 М Трис, рН 7,3, при температуре 37 С. Вводится свиной трипсин (75 г). Раствор тщательно смешивается и время от времени перемешивается в течение 120 мин при 37 С. В этот момент времени реакция прекращается в результате ввода этой смеси в 137 мл 0,05 н. НС 1. Весь этот раствор вводится в колонку размерами 21250 мм с С-8 , и продукты элюируются при небольшом ацетонитриловом градиенте в буферном растворе 0,1 М одноосновного фосфата натрия, рН 2. Соответствующие фракции, которые определены методом аналитической , сливаются, в два раза разбавляются водой и вводятся в колонку 25300 мм с С-18 . Желаемый белок элюируется из колонки ацетонитрильным градиентом в 0,05 -ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются с получением выхода 59 мг. Указанная структура подтвер 7 3225 1 ждается аминокислотным анализом (табл. 1) и масс-спектроскопией .5765,7 (теоретически 5765,6). Пример 4. Человеческий инсулин(В 29),(В 28). Свиной -октапептидный инсулин (290 мг) и систематический октапептид (310 мг) смешиваются в 10 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,4 бутандиола и 1 часть буферного раствора 0,25 М Трис, рН 7,3. Вводится свиной трипсин (60 мг). Раствор тщательно смешивается и время от времени перемешивается в течение 60 мин при 37 С. В этот момент времени реакция прекращается за счет ввода смеси в 90 мл 0,05 н. НС 1. Весь раствор вводится в колонку размерами 21250 мм с С-8 , и продукты элюируются в небольшом ацетонитрильном градиенте в буферном растворе 0,1 М одноосновного фосфата натрия, рН 2. Соответствующие фракции, которые определены методом аналитической , сливаются, в два раза разбавляются водой и вводятся в колонку размерами 10250 мм с С-8 . Желаемый белок элюируется из колонки с использованием ацетонитрильного градиента в 0,5 -ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются с получением выходы 43 мг. Указанная структура подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией .5752,3 (теоретически 5751,6). Пример 5. Человеческий инсулин(В 29),(В 28). Свиной -октапептидный инсулин (290 мг) и синтетический октапептид (310 нг) смешивают в 10 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,4-бутандиола и 1 часть буферного раствора 0,25 М Трис, рН 7,3, при 37 С. Вводится свиной инсулин (60 мг). Раствор смешивают и время от времени тщательно перемешивается в течение 60 мин при 37 С. В этот момент времени реакция прекращается в результате ввода этой смеси в 90 мл 0,05 н. НС 1. Весь раствор вводится в колонку размерами 21250 мм с С-8, и продукты элюируются в небольшом ацетонитрильном градиенте в буферном растворе 0,1 М одноосновного фосфата натрия, Н 2. Соответствующие фракции, которые определены аналитической , сливаются, в два раза разбавляются водой и вводятся в колонку 10250 мм с С-8 . Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5 -ной трифторуксусной кислоте, фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются, и получается 103 мг продукта. Указанная структура подтверждается аминокислотным анализом (табл. 1) и масс-спектрометрическим анализом .5836,1 (теоретически 5836,7). Пример 6. Человеческий инсулин(В 29),(В 28). Свиной -октапептидный инсулин (409 мг) и синтетический октапептид (398 мг) смешиваются в 14 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,4 бутандиола и 1 часть буферного раствора 0,25 М Трис, рН 7,9, при 37 С. Вводят свиной трипсин (81 мг). Раствор смешивают и время от времени тщательно перемешивают в течение 120 мин при 37 С. В этот момент времени реакция прекращается за счет ввода смеси в 136 мл 0,5 н. НС 1. Весь раствор вливается в колонку 21250 мм с С-8 , и продукты элюируются в небольшом ацетонитрильном градиенте в буферном растворе 0,2 М одноосновного фосфата натрия, рН 2. Соответствующие фракции, которые определены посредством аналитической , сливаются, в два раза разбавляются водой и вводятся в колонку размерами 25300 мкм с С-18 . Желаемый белок элюируется из колонки ацетонитрильным градиентом в 0,5 -ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются, в результате получается 56 мг продукта. Указанная структура подтверждается аминокислотным аналогом (табл. 1) и масс-спектроскопией .5794,7 (теоретически 5794,6). Пример 7. Человеческий инсулин(В 29),(В 28). Свиной -октапептидный инсулин (400 мг) и синтетический октапептид (388 мг) смешиваются в 13 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,4 бутандиола и 1 часть буферного раствора 0,25 М Трис, рН 7,3 при 37 С. Вводится свиной трипсин (78 мг). Раствор тщательно смешивается и время от времени перемешивается в течение 120 мин при 37 С. Реакция прекращается в данный момент времени путем ввода этой смеси в 137 мл 0,05 н. НС 1. Весь раствор вводится в колонку 21250 мм с С-8 , и продукты элюируются небольшим ацетонитрильным градиентом в буферном растворе 0,1 М одноосновного фосфата натрия рН 2. Соответствующие фракции, которые определены аналитической , сливаются, разбавляются в четыре раза водой и вводятся в колонку размерами 25300 мм с С-8 . Желаемый белок элюируется из колонки ацетонитрильным градиентом в 0,1 -ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливают и лиофилизируются, в результате получается 85 мг продукта. Указанная структура подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией .5795,7 3225 1 Для получения сырой пептидной смолы используется пептидный синтезатор 430 А(включая модификацию 1,4). Используется 0,5 ммоль исходной твердофазной смолы (2 смолы) (0,72 ммоль/г 0,705 г). Все используемые аминокислоты представляют собой защищенные ВОС, за исключением глутаминовой кислоты, аспарагиновой кислоты и гистидина, все аминокислоты используются в том виде, как они получены (то есть в упаковках, поставляемых фирмой. каждая упаковка содержит примерно 2 ммоль защищенной аминокислоты). Глутаминовая кислота, аспарагиновая кислота и гистидин являются промышленно получаемыми кислотами, они заключаются в упаковки таким образом, что каждая упаковка содержит примерно 2 ммоль желаемой защищенной аминокислоты. Сырая пептидиловая смола высушивается в вакууме при комнатной температуре в течение ночи и ее вес сопоставляется с весом исходной смолы для гарантии нужного привеса. Небольшая часть образца подвергается аминокислотному анализу для гарантии того, что аминокислоты вводятся в правильных пропорциях. Пептид отщепляется от пептидиловой смолы, и защита боковой цепи удаляется в результате перемешивания в течение примерно 1 ч при 0 С в растворе 10 частей (об./вес.)(содержащего 5 об./об. паратиокрезола и 5 об./об. метакрезола) с 1 частью пептидиловой смолы. После удаления наибольшей частипри вакуумной отгонке пептид осаждается в простом этиловом эфире. После нескольких промывок простым этиловым эфиром с последующей вакуумной фильтрацией пептид растворяется примерно в 120 мл 8 М гуанидина в НС 1, рН 11, содержащего 0,1 моль Трис, 35 мг/мл 23 и 25 мг/мл 249. Величина рН раствора доводится до 8,8 посредством 5 н. , и раствор интенсивно перемешивается в течение 3 ч при комнатной температуре. Полученный -сульфонированный пептидный раствор вводится в колонку 5215 см с-25 при комнатной температуре. Образец элюируется со скоростью 21 мл/мин при комнатной температуре с использованием 50 ммоль бикарбоната аммония. Выходящий из колонки продукт анализируется (при 276 нм). Фракции по 25 мл собираются и сливаются, и собранный продукт из желаемых фракций дополнительно очищается путем жидкостной хроматографии высокого разрешения , как описано ниже. Слитный продукт, состоящий из желаемых фракций, вводится в хроматографическую колонку 2,530 см сС-8, 9-12 мкм, и элюирование осуществляется с использованием линейного градиента с увеличивающейся концентрацией ацетонитрила в 100 ммоль бикарбоната аммония при комнатной температуре (2,6 мл/мин). Выходящий из колонки продукт анализируется при 280 нм. Фракции (по 25 мл) собираются и сливаются. Отобранные фракции подвергаются анализу методом аналитическойдля определения того,какие фракции сохраняются. Желаемые фракции сливаются, лиофилизируются и используются в указанной ниже комбинации с А-цепью. В. Комбинация В-цепи человеческого инсулина(В 29),(28),(10) с А-цепью человеческого инсулина. Комбинация А- и В-цепей осуществляется согласно процедуре, описанной в работеи др., см. выше. Два грамма -сульфоната А-цепи, образованного от рекомбинантной ДНК, и 400 мг синтетического сульфоната В-цепи(В 29),(10),(28) каждого растворяются в 200 мл и 40 мл соответственно 0,1 М буферного раствора глицерина при комнатной температуре, величина рН каждого доводится до 10,5 посредством 5 н.и затем каждый охлаждается до 5 С. Приготавливается раствор дитиотретиола(ДТТ) концентрацией 15,5 мг/мл в 0,1 М буферном растворе глицина при комнатной температуре, величина рН доводится до 10,5 посредством 5 н. , и затем раствор охлаждается до 5 С. Растворы А-цепи и В-цепи смешиваются, и затем в эту смесь быстро вводится 15,9 мл раствора ДТТ(/ 3). Реакционный раствор перемешивается при 4 С в открытых 200-миллилитровых стеклянных пробирках для центрифугирования в течение 19,6 ч при 4 С. Вводится ледяная уксусная кислота (129 мл), и раствор выстаивается при 4 С в течение 1 ч. Полученная смесь с выпавшим осадком центрифугируется в течение 30 мин со скоростью 2000 об/мин при 4 С. Поверхностный слой смешивается с 292 мл милливоды и 73 мл ацетонитрила, и эта смесь дополнительно очищается посредствомс обратимой фазой (с использованием колонки 2,530 см с 18 с элюированием при комнатной температуре со скоростью 2,6 мл/мин с использованием линейного ингредиента с увеличивающейся концентрацией ацетонитрила в 0,1 М 24, рН 2,1). Выходящий из колонки продукт анализируется при 280 нм. Выбранные фракции анализируются методом аналитической, и желаемые фракции сливаются и разбавляются в два раза 0,1 -ным водным раствором трифторуксусной кислоты , затем слитый продукт вводится в хроматографическую колонку (0,125 см) с , и осуществляется элюирование линейным градиентом с увеличивающейся концентрацией ацетонитрила в 0,1 -ном водном растворе . Выходящий из колонки продукт анализируется при 280 нм. Выбранные фракции анализируются методом аналитической , и желаемые фракции сливаются и повторно очищаются с использованием той же колонки и в тех же условиях, что указаны выше, при несколько отличающемся градиенте ацетонитрила. Соответствующие фракции сливаются и лиофилизируются, в результате получается 65 мг аналога инсулина со степенью чистоты более чем 93 в результатес обра 9 3225 1 тимой фазой. Указанная структура подтверждается аминокислотным анализом (табл. 1) и методом массспектроскопии .5786,1 (теоретически 5786,7). Пример 9. Человеческий инулин(В 29), Суа (В 28). Осуществляется окисление надмуравьиной кислоты с целью превращения цистеина откапептида в форму цистеиновой кислоты. Синтетический октапептид Ту (363 мг) растворяется в 36 мл свежеприготовленной надмуравьиной кислоты в охлажденной льдом колбе, и раствор осторожно перемешивается в течение 1 ч. Окисленный материал разбавляется в десять раз водой и лиофилизируется. В данном полусинтезе используется этот лиофилизат. Свиной -октапептидный инсулин (222 мг) и синтетический октапептид (225 мг) смешивают в 18 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,4-бутандиола и 1 часть буферного раствора 0,25 М Трис, рН 7,3, при 37 С. Вводится свиной трипсин (45 мг). Раствор тщательно смешивается и время от времени перемешивается в течение 120 мин при 37 С. В этот момент времени реакция прекращается в результате ввода этой смеси в 242 мл 0,05 н. НС. Весь раствор вводится в колонку 21250 см с С-8 , и продукты элюируются слабым ацетонитрильным градиентом в буферном растворе 0,1 М одноосновного фосфата натрия, рН 2. Подходящие фракции, которые определены методом аналитической , сливаются, разбавляются в два раза водой и вводятся в колонку 25300 мм с С-18 . Обессоленный белок элюируется из колонки с использованием ацетонитрильного градиента в 0,5 -ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются, в результате получается выход продукта 16 мг. Указанная структура продукта подтверждается аминокислотным анализом (табл. 1) и методом массспектроскопии .5831,5 (теоретически 5831,7). Пример 10. Человеческий инсулин Р (В 29),(В 28). Свиной -октапептидный инсулин (290 мг) и синтетический октапептид (310 мг) смешиваются в 10 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,4 бутандиола и 1 часть буферного раствора 0,25 М Трис, рН 7,3, при 37 С. Вводится свиной трипсин (60 мг). Раствор тщательно смешивается и время от времени перемешивается в течение 60 мин при 37 С. В этот момент времени реакция прекращается в результате ввода смеси в 90 мл 0,05 н. НС. Весь раствор вводится в колонку 21250 мм с С-8 , и продукты элюируются с использованием слабого ацетонитрильного градиента в буферном растворе 0,1 М одноосновного фосфата натрия, рН 2. Подходящие фракции, которые определены путем аналитической , сливаются, разбавляются в два раза водой, вводятся в колонку размерами 10250 мм с С-8 . Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5 -ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина,сливаются и лиофилизируются, получается выход продукта 87 мг. Указанная структура подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией .5809,4 (теоретически 5808,6). Пример 11. Человеческий инсулин(В 29),(В 28). Свиной -октапептидный инсулин (402 мг) и синтетический октапептид (398 мг) смешиваются в 14 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,4 бутандиола и 1 часть буферного раствора 0,25 М Трис, при 37 С. Вводится свиной трипсин (80 мг). Раствор тщательно смешивается и время от времени перемешивается в течение 120 мин при 37 С. В этот момент времени реакция прекращается путем ввода этой смеси в 136 мл 0,05 н. НС. Весь раствор вводится в колонку 21250 мм с С-8 , и продукты элюируются в слабом ацетонитрильном градиенте в буферном растворе 0,1 М одноосновного фосфата натрия, рН 2. Подходящие фракции, которые определены путем аналитической , сливаются, разбавляются в четыре раза водой и вводятся в колонку 25300 мм с С-18 . Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5 -ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются, в результате получается выход продукта 59 мг. Данная структура подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией .5809,6(теоретически 5809,6). Пример 12. Человеческий инсулин(В 29),(В 28). Свиной -октапептидный инсулин (412 мг) и синтетический октапептид (376 мг) смешиваются в 13 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,4 бутандиола и 1 часть буферного раствора 0,25 М Трис, рН 7,3. Вводят свиной трипсин (79 мг). Раствор тщательно смешивается и время от времени перемешивается в течение 180 мин при 37 С. В этот момент времени реакция прекращается путем ввода смеси в 147 мл 0,05 н. НС. Весь раствор вводится в колонку 21250 мм с С-8 , и продукты элюируются слабым ацетонитрильным градиентом в 0,1 М буферном растворе одноосновного фосфата натрия, рН 2. Подходящие фракции, которые определены аналитической , сливаются, разбавляются в четыре раза водой и вводятся в колонку 25300 мм с С-18 . Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,1 -ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются, в результате получается 11 мг продукта. Данная структура про 10 3225 1 дукта подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией .5737,2 (теоретически 6537,6). Пример 13. Человеческий инсулин(В 29),(В 28). Свиной -октапептидный инсулин (400 мг) и синтетический октапептид (398 мг) смешиваются в 13 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,4 бутандиола и 1 часть буферного раствора 0,25 М Трис, рН 7,3, при 37 С. Вводится свиной трипсин (79 мг). Раствор тщательно смешивается и время от времени перемешивается в течение 120 мин при 37 С. В этот момент времени реакция прекращается путем ввода смеси в 237 мл 0,05 н. НС 1. Весь раствор вводится в колонку 21250 мм с С-8 , и продукты элюируются в слабом ацетонитрильном градиенте в буферном растворе 0,1 М одноосновного фосфата натрия, рН 2. Подходящие фракции, которые определены аналитической , сливаются, разбавляются в четыре раза водой, вводятся в колонку 25300 мм с С-18 . Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,1 -ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются, в результате получается выход продукта 79 мг. Данная структура подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией .5816,9 (теоретически 5817,7). Пример 14. Человеческий инсулин(В 29),(В 28). Свиной -октапептидный инсулин (409 мг) и синтетический октапептид (398 мг) смешиваются в 13 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,4 бутандиола и 1 часть буферного раствора 0,25 М Трис, рН 7,3, при 37 С. Вводят свиной трипсин (81 мг). Раствор тщательно смешивается и время от времени перемешивается в течение 120 мин при 37 С. В этот момент времени реакция прекращается путем ввода смеси в 136 мл 0,05 н. НС. Весь раствор вводится в колонку 21250 мм с С-8 , и продукты элюируются в слабом ацетонитрильном градиенте в буферном растворе 0,1 М одноосновного фосфата натрия, рН 2. Подходящие фракции, определяемые аналитической , сливаются, разбавляются в два раза водой и вводятся в колонку 25300 мм с С-18 . Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5 -ной трифторуксусной кислоте. Фракции, содержащие очищенный инсулиновый аналог,сливаются и лиофилизируются, в результате получается выход продукта 57 мг. Данная структура подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией .5793,7 (теоретически 5793,7). Пример 15. Человеческий инсулин(В 29), (28). Свиной -октапептидный инсулин (418 мг) и синтетический октапептид (410 мг) смешиваются в 14 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,4 бутандиола и 1 часть буферного раствора 0,25 М Трис, рН 7,3, при 37 С. Вводится свиной трипсин (83 мг). Раствор тщательно смешивается и время от времени перемешивается в течение 120 мин при 37 С. В этот момент реакция прекращается путем ввода этой смеси в 136 мл 0,05 н. НС. Весь раствор вводится в колонку 21250 мм с С-8 , и продукты элюируются в слабом ацетонитрильном градиенте в 0,1 М буферном растворе одноосновного сульфата натрия, рН 2. Соответствующие фракции, определяемые аналитической , сливаются, разбавляются в четыре раза водой и вводятся в колонку 25300 мм с С-18 . Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5-ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются, в результате получается выход продукта 74 мг. Структура этого продукта подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией .5793,8 (теоретически 5793,7). Пример 16. Человеческий инсулин(В 29),(В 28). Свиной -октапептидный инсулин (290 мг) и синтетический октапептид (310 мг) смешиваются в 10 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,4 бутандиола и 1 часть буферного раствора 0,25 М Трис, рН 7,3, при 37 С. Вводят свиной трипсин (60 мг). Раствор тщательно смешивается и время от времени перемешивается в течение 60 мин при 37 С. В этот момент реакция прекращается путем ввода смеси в 90 мл 0,05 н. НС. Весь раствор вводится в колонку 21250 мм с С-8 , и продукты элюируются в слабом ацетонитрильном градиенте в 0,1 М буферном растворе одноосновного фосфата натрия, рН 2. Подходящие фракции, которые определены аналитической , сливаются, разбавляются в два раза водой и вводятся в колонку 10259 мм с С-8 . Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5 -ной трифторуксусной кислоте. Фракции, содержащие инсулиновый аналог, сливаются и лиофилизируются, в результате получается выход продукта 54 мг. Указанная структура подтверждается аминокислотным анализом (табл. 1) и массспектроскопией .5794,6 (теоретически 5793,7). Пример 17. Человеческий инсулин - (В 29). Свиной -октапептидный инсулин (392 мг) и синтетический октапептид (387 мг) смешиваются в 23,5 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,411 3225 1 бутандиола и 1 часть буферного раствора 0,25 М Трис, рН 7,3, при 37 С. Вводят свиной трипсин (78 мг). Раствор тщательно смешивается и время от времени перемешивается в течение 120 мин при 37 С. В этот момент реакция прекращается путем ввода смеси в 136,5 мл 0,05 н. НС. Весь раствор вводится в колонку 21250 мм с С-8 , и продукты элюируются в слабом ацетонитрильном градиенте в 0,1 М буферном растворе одноосновного сульфата натрия, рН 2. Подходящие фракции, определяемые аналитической , сливаются, разбавляются в четыре раза водой и вводятся в колонку 25300 мм с С-18 . Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5 -ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина,сливаются и лиофилизируются, в результате получается 94 кг продукта. Данная структура подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией .5809,0 (теоретически 5808,7). Пример 18. Человеческий инсулин(В 29),(В 28). Свиной -октапептидный инсулин (350 мг) и синтетический октапептид (366 мг) смешиваются в 12 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,4 бутандиола и 1 часть буферного раствора 0,25 М Трис, рН 7, при 37 С. Вводится свиной трипсин (71 мг). Раствор тщательно смешивается и время от времени перемешивается в течение 120 мин при 37 С. В данный момент времени реакция прекращается путем ввода смеси в 118 мл 0,05 н. . Весь раствор вводится в колонку 21250 мм с С-8 , и продукты элюируются в слабом ацетонитрильном градиенте в 0,1 М буферном растворе одноосновного фосфата натрия, рН 2. Подходящие фракции, определяемые аналитической , сливаются, разбавляются в четыре раза водой и вводятся в колонку 25300 мм с С-18 . Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,1 -ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина,сливаются и лиофилизируются, в результате получается 72 мг продукта. Указанная структура подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией .5811,0 (теоретически 5811,7). Пример 19. Человеческий инсулин(В 29), О (В 28). Свиной -октапептидный инсулин (290 мг) и синтетический октапептид (310 мг) смешиваются в 10 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,4-бутандиола и 1 часть буферного раствора 0,25 М Трис, рН 7,3, при 37 С. Вводится свиной трипсин (60 мг). Раствор тщательно смешивается и время от времени перемешивается в течение 90 мин при 37 С. В этот момент времени реакция прекращается путем ввода данной смеси в 90 мл 0,05 н. НС. Весь раствор вводится в колонку 21250 мм с С-8 , и продукты элюируются в слабом ацетонитрильном градиенте в 0,1 М буферном растворе одноосновного фосфата натрия, рН 2. Подходящие фракции, определяемые аналитической , сливаются, разбавляются в четыре раза водой и вводятся в колонку 10250 мм с С-8 . Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5 -ной трифторуксусной кислоте. Фракции, содержащие очищенный инсулиновый аналог, сливаются и лиофилизируются, в результате получается 89 мг продукта. Структура продукта подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией .5795,2 (теоретически 5794,7). Пример 20. Человеческий инсулин(В 29),(В 28). Свиной -октапептидный инсулин (290 мг) и синтетический октапептид (310 мг) смешивают в 10 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,4-бутандиола и 1 часть буферного раствора 0,25 М Трис, рН 7,3, при 37 С. Вводится свиной трипсин (60 мг). Раствор тщательно смешивается и время от времени перемешивается в течение 80 мин при 37 С. В этот момент времени реакция прекращается путем ввода смеси в 90 мл 0,05 н. НС. Весь раствор вводится в колонку 21250 мм с С-8 , и продукты элюируются в слабом ацетонитрильном градиенте в 0,1 М буферном растворе одноосновного фосфата натрия, рН 2. Подходящие фракции, определяемые аналитической , сливаются, разбавляются в четыре раза водой и вводятся в колонку 25300 мм с С-18 . Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,1 -ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина,сливаются и лиофилизируются, в результате получается 17 мг продукта. Структура продукта подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией .5827,9 (теоретически 5827,7). Пример 21. Человеческий инсулин(В 29). Свиной -октапептидный инсулин (339 мг) и синтетический октапептид (363 мг) смешивают в 9 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,4-бутандиола и 1 часть буферного раствора 0,25 М Трис, рН 7,3 при 37 С. Вводится свиной трипсин (70 мг). Раствор тщательно смешивается и время от времени перемешивается в течение 80 мин при 37 С. Данная реакция в этот момент времени прекращается путем ввода смеси в 108 мл 0,05 н. НС. Весь раствор вводится в колонку 10250 мм с С-8 , и продукты элюируются слабым ацетонитрильным градиентом в 0,1 М буферном растворе одноосновного фосфата натрия, рН 2. Подходящие фракции, которые определены аналитической , сливаются, разбавляются два раза водой и вводятся в колонку 10250 мм с -8 . Обессоленный белок элюируется из колонки ацето 12 3225 1 нитрильным градиентом в 0,5 -ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются, в результате получается 97 мг продукта. Структура подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией .5778,6 (теоретически 5777,6). Пример 22. Человеческий инсулин(В 29),(В 28). Свиной -октапептидный инсулин (412 мг) и синтетический октапептид (390 мг) смешиваются в 13 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,4 бутандиола и 1 часть буферного раствора 0,25 М Трис, рН 7,3, при 37 С. Вводится свиной трипсин (80 мг). Раствор тщательно смешивается и время от времени перемешивается в течение 120 мин при 37 С. В данный момент времени реакция прекращается путем ввода смеси в 137 мг 0,05 н. НС. Весь раствор вводится в колонку 21250 мм с -8 , и продукты элюируются в слабом ацетонитрильном градиенте в 0,1 М буферном растворе одноосновного фосфата натрия, рН 2. Подходящие фракции, определяемые аналитической , сливаются, разбавляются в два раза водой и вводятся в колонку 25 х 300 мм с С-18 . Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5 -ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, собираются и лиофилизируются с получением выхода продукта 37 мг. Структура продукта подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией .5768,1 (теоретически 5767,6). Пример 23. Человеческий инсулин(В 29),(В 28). Свиной -октапептидный инсулин (437 мг) и синтетический октапептид (420 мг) смешиваются в 14,5 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,4 бутандиола и 1 часть буферного раствора 0,25 М Трис, рН 7,3, при 37 С. Вводят свиной трипсин (86 мг). Раствор тщательно смешивается и время от времени перемешивается в течение 120 мин при 37 С. В этот момент времени реакция прекращается путем ввода смеси в 135,5 мл 0,05 н. НС. Весь раствор вводится в колонку 21 х 250 мм с -8 , и продукты элюируются небольшим ацетонитрильным градиентом в буферном растворе 0,1 М одноосновного фосфата натрия, рН 2. Подходящие фракции, определяемые аналитически , сливаются, в два раза разбавляются водой и вводятся в колонку 25 х 300 мм с С-18 . Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5 -ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются, получается выход продукта 78 мг. Данная структура подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией .5781,9 (теоретически 5781,6). Пример 24. Человеческий инсулин , (В 29), Тр (В 28). Свиной -октапептидный инсулин (310 мг) и синтетический октапептид (325 мг) смешиваются в 10,5 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,4 бутандиола и 1 часть буферного раствора 0,28 М Трис, рН 7,3, при 37 С. Вводится свиной трипсин (64 мг). Раствор тщательно смешивается и время от времени перемешивается в течение 120 мин при 37 С. В этот момент времени реакция прекращается путем ввода смеси в 140 мл 0,05 н. НС. Весь раствор вводится в колонку 21 х 250 мм с -8 , и продукты элюируются слабым ацетонитрильным градиентом в 0,1 М буферном растворе одноосновного фосфата натрия, рН 2. Подходящие фракции, которые определены аналитической , сливаются, в два раза разбавляются водой и вводятся в колонку размерами 25 х 300 мм с С-18 . Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5 -ной трифторуксусной кислоте. Фракции, содержащие очищенный инсулин, сливаются и лиофилизируются, в результате получается выход продукта 47 мг. Данная структура подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией .5866,2 (теоретически 5866,7). Пример 25. Человеческий инсулин(В 29), Ту (В 28). Свиной -октапептидный инсулин (391 мг) и синтетический октапептид (400 мг) смешиваются в 13 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,4 бутандиола и 1 часть буферного раствора 0,25 М Трис, рН 7,3, при 37 С. Вводится свиной трипсин (79 мг). Раствор тщательно смешивается и время от времени перемешивается в течение 120 мин при 37 С. В этот момент времени реакция прекращается путем ввода смеси в 137 мл 0,05 н. НС. Весь раствор вводится в колонку размерами 21 х 250 мм с С-8 , и продукты элюируются слабым ацетонитрильным градиентом в буферном растворе 0,1 М одноосновного фосфата натрия, рН 2. Подходящие фракции, которые определены аналитической , сливаются, разбавляются в два раза водой и вводятся в колонку размерами 25 х 300 мм с С-18 . Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5 -ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются, в результате получается выход продукта 30 мг. Данная структура подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией .5843,7 3225 1 1 часть буферного раствора 0,25 М Трис, рН 7,3, при 37 С. Вводится свиной трипсин (78 мг). Раствор тщательно смешивается и время от времени перемешивается в течение 120 мин при 37 С. В этот момент времени реакция прекращается путем ввода смеси в 238 мл 0,05 н. НС. Весь раствор вводится в колонку 21 х 250 мм с С-8 , и продукты элюируются слабым ацетонитрильным градиентом в 0,1 М буферном растворе одноосновного фосфата натрия, рН 2. Подходящие фракции, которые определены аналитической , сливаются, разбавляются в четыре раза водой и вводятся в колонку размерами 25 х 300 с С-18 . Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,1 -ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются, в результате получается выход продукта 74 мг. Структура продукта подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией .5780,0 (теоретически 5799,6). Пример 27. Человеческий инсулин(В 29),(В 28). Свиной -октапептидный инсулин (292 мг) и синтетический октапептид (279 мг) смешиваются в 10 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,4 бутандиола и 1 часть буферного раствора 0,25 М Трис, рН 7,3, при 37 С. Вводится свиной трипсин (57 мг). Раствор тщательно смешивается и время от времени перемешивается в течение 120 мин при 37 С. В этот момент времени реакция прекращается путем добавления смеси в 240 мл 0,05 н. НС. Весь раствор вводится в колонку 21 х 250 мм с С-8 , и продукты элюируются слабым ацетонитрильным градиентом в буферном растворе 0,1 М одноосновного фосфата натрия, рН 2. Подходящие фракции, которые определены аналитической , сливаются, разбавляются в два раза водой и вводятся в колонку размерами 25 х 300 мм с С-18 . Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5 -ной трифторуксусной кислоте. Фракции, содержащие очищенный инсулиновый аналог, сливаются и лиофилизируются, в результате достигается выход продукта 51 мг. Структура продукта подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией .5870,0(теоретически 5779,6). Пример 28. Осуществляя процедуры так, как описано в данной заявке, получают нижеследующие дополнительные аналоги инсулина человеческий инсулин (1), (28), Ро(В 29). Пример 29. Человеческий инсулин(В 29),(В 28). А. Построение плазмиды рСР 126. 1. Выделение плазмиды рКС 283. Лиофилы .К 12 ВЕ 1201/рКС 283 получены из Северной Региональной Научно-исследовательской Лаборатории, Пеория, Иллинойс 61604, где они имеют официальный номерВ-15830. Эти лиофилы декантируются в пробирки, содержащие 10 мл среды(10 г Бакто-триптона, 5 г Бакто-дрожжевого экстракта и 10 гна литр рН доводится до 7,5), и инкубируются в течение 2 ч при 32 С, в течение этого времени культуры доводятся до концентрации 50 мкг/мл в ампициллине и затем инкубируются при 32 С в течение ночи. Клетки .К 12 ВЕ 1201/рКС 283 выращивают при 32 С, поскольку они содержат температурочувствительный подавляющий ген, интегрированный в клеточную ДНК. При использовании в процедурах выделения плазмиды (как описано в нижеследующих примерах) клеток, которые содержат подавляющий ген лямбда р дикого типа или не содержат промотора лямбда р, температура инкубации составляет 37 С. Небольшая часть выращенной за ночь культуры помещается в чашки с -агаром (средас 15 г/л Бакто-агара), содержащие 50 мкг/мл ампициллина, так, чтобы получился один единственный изолят колонки .К 12 ВЕ 1201/рКС 283. Эта единственная полученная колония инокулируется в 10 мл среды , содержащей 50 мкг/мл ампициллина, и инкубируется в течение ночи при 32 С при интенсивном встряхивании. 10 мл ночной культуры инокулируется в 500 мл среды , содержащей 50 мкг/мл ампициллина, и инкубируется при 32 С с интенсивным встряхиванием до тех пор, пока культура не достигает стационарной фазы. Принята нижеследующая процедура, описанная в работеи др., 1982 г.,. Клетки собираются путем центрифугирования с ускорением 4000 в течение 10 мин при 4 С, и поверхностный слой удаляется. Клеточный осадок промывается в 100 мл охлажденного льдом буферного раствора 14 3225 1(0,1 моль , 10 ммоль Трис-, рН 7,8, и 1 ммоль ЭДТА). После промывки клеточный осадок повторно суспензируется в 10 мл раствора 1 (50 ммоль глюкозы, 25 ммоль Трис-, рН 8, и 10 ммоль ЭДТА),содержащего 5 мг/мл лизозима, и оставляется при комнатной температуре в течение 10 мин. Затем в обработанные лизозимом клетки вводится 20 мл раствора 2 (2 н.и 1), и раствор подвергается осторожному инверсионному перемешиванию. Смесь инкубируется на льду в течение 10 мин. 15 мл охлажденного льдом 5 М ацетата калия, рН 4,8, вводятся в лизированную клеточную смесь, и раствор подвергается инверсионному перемешиванию. Приготавливается 5 М раствор ацетата калия путем ввода 11,5 мл ледяной уксусной кислоты в 28,5 мл воды и 60 мл 5 М ацетата калия, полученный раствор является 3 М в отношении концентрации калия и 5 М в отношении концентрации ацетата. Лизированная клеточная смесь центрифугируется в центрифуге Бекмана 27 (или его эквивалента) со скоростью 20000 об/мин в течение 20 мин при температуре 4 С. Клеточная ДНК и клеточные продукты распада образуют осадок на дне пробирки. Извлекается примерно 36 мл поверхностного слоя и вводится 0,6 объема изопропанола, осуществляется перемешивание, и полученный раствор выдерживается при комнатной температуре в течение 15 мин. Плазмидная ДНК извлекается путем центрифугирования с ускорением 12000 в течение 3 мин при комнатной температуре. Поверхностный слой декантируется, и осадок ДНК промывается 70 -ным этанолом при комнатной температуре. Затем этанольная промывка декантируется, и осадок высушивается в эксикаторе. Этот осадок снова суспендируется в 8 мл буферного раствора ТЕ (10 мл Трис-, рН 8, и 1 мл этилендиаминтетраацетата (ЭДТА). В раствор ДНК вводится 8 г СаС 2, вводится примерно 0,8 мл раствора этилиденбромида в воде на каждые 10 мл раствора СаС 2-ДНК. Конечная плотность этого раствора составляет примерно 1,55 г/мл, и концентрация этилиденбромида составляет примерно 600 мкг/мл. Данный раствор подается в пробирку для центрифугирования типа Бекмана 50, наполняется сверху парафиновым маслом, уплотняется и центрифугируется со скоростью 45000 об/мин в течение 24 ч при 20 С. После центрифугирования в обычном свете наблюдаются два слоя ДНК. После удаления крышки с пробирки нижний слой ДНК удаляется посредством шприца с гиподермической иглой 21, вставляемой в пробирку для центрифугирования сбоку. Этиленбромид удаляется путем нескольких экстракций 1-бутанолом, насыщенным водой. 2 удаляется путем диализа с ТЕ-буфером. После экстракций буферным раствором фенола, а затем хлороформом ДНК осаждается, промывается 70 -ным этанолом и высушивается. Получается примерно 1 мг плазмиды рКС 283, и она выдерживается при 4 С в буферном растворе ТЕ концентрацией примерно 1 мкг/мкл. Ограничительная точка и функциональная карта плазмиды рКС 283 представлены на фиг. 2. 2. Построение плазмиды рКС 283 РХ. Примерно 10 мкл ДНК плазмиды рКС 283, приготовленной как описано в примере 1, смешивается с 20 мкл 10 Х среднесолевого ограничительного буфера (500 ммоль , 100 ммоль Трис-, рН 7,5, 100 ммоль 2 и 10 ммоль ДТТ), 20 мкл(1 мг/мл), 5 мкл ограничительного фермента 11 ( 50 ед., как определено в лаборатории, из которой все эти ограничительные ферменты получены) и 145 мкл воды, и эта реакционная смесь инкубируется при 37 С в течение 2 ч. Реакции ограничительного фермента, завершаются обычным образом путем экстракции фенолом, а затем хлороформом, после чего происходит осаждение ДНК, который промывается этанолом и снова суспензируется в буферном растворе ТЕ. После завершения выпаривания с , как описано выше, вываренная с 11 плазмидная рКС 283 ДНК осаждается и затем снова суспензируется в 5 мкл буферного раствора ТЕ. Примерно 500 пмоль связывающих звеньев(53) киназируются в смеси, содержащей 10 мкл 5 Х киназного буферного раствора (300 ммоль Трис-, рН 7,8, 50 ммоль 2 и 25 ммоль ДТТ), 5 мкл 5 М АТР, 24 мкл Н 2, 0,5 мкл полинуклеотидокиназы Т 4 (примерно 2,5 единиц, как определено в ), 5 мкл 1 мг/мли 5 мкл 10 мМ спермидина, путем инкубирования этой смеси при 37 С в течение 30 мин. Примерно 12,5 мкл киназированных связующих звеньев вводятся в 5 мкл вываренных сДНК плазмиды рКС 283, и затем 2 мкл 10 Х буферного раствора лигазы (300 ммоль Трис-, рН 7,6, 100 ммоль 2 и 50 ммоль ДТТ), 2,5 мклконцентрацией 1 мг/мл, 7 мкл 5 мМ АТР, 2,5 мкл лигазы Т 4 (ДНК примерно 2,5 ед., как определено - ), 2,5 мкл 10 мМ спермидина и 3 мкл воды вводятся в указанную ДНК. Полученная реакционная смесь сшивки инкубируется при 4 С в течение ночи. После реакции сшивки состав реакционной смеси регулируется таким образом, чтобы получился высокосолевой буферный раствор(0,1 моль , 0,05 моль Трис-, рН 7,5, 10,0 ммоль 2 и 1 ммоль ДТТ). В смесь вводится примерно 10 мкл (100 ед.) ограничительного фермента , и полученная реакционная смесь инкубируется при 37 С в течение 2 ч. Реакция завершается, и вываренная сДНК осаждается, повторно суспензируется и сшивается, как описано выше, с той разницей, что никаких связывающих звеньевне вводится в смесь сшивки. Сшитая ДНК состоит из желаемой плазмиды рКС 283 РХ. Ограничительная точка и функциональная карта плазмиды рКС 283 РХ представлены на фиг. 3. 3. Построение .К 12 МО /рКС 283 РХ. 15.К 12 МОможет быть получена из Северных Региональных Научных Лабораторий в лиофилизированной форме под официальным номеромВ-15993. .К 12 МО включает подавляющий ген лямбдаданного типа, так что транскрипции от гибридного промотора -, отвечающего данному изобретению, не происходит в клетках .К 12 МО . Осуществляется выделение лиофилов и перестроенных одиночных колоний МО , и приготавливается 10 мл ночной культуры клеток МОв основном согласно процедуре, описанной в примере 29 А 1, с той разницей, что температура инкубации составляет 37 С и в среде роста не используется никакого ампициллина. 50 мкл ночной культуры используется для инкуляции 5 мл среды , которая также содержит 2 ммоль 4 и 10 ммоль 2. Данная культура инкубируется при 37 С в течение ночи при интенсивном встряхивании. На следующее утро культура разбавляется до 200 мл средой , содержащей 10 ммоль 4 и 10 ммоль 2. Разбавленная культура инкубируется при 37 С при интенсивном встряхивании до тех пор, пока поглощение при 550 мм (А 550) не составит примерно 0,5, что показывает клеточную плотность примерно 110 клеток/мл. Эта культура охлаждается в течение 10 мин в ледяной водяной бане, и затем клетки извлекаются путем центрифугирования при ускорении 4000 в течение 10 мин при 4 С. Клеточный осадок на дне пробирки снова суспензируется в 100 мл холодного 10 мМ 4 и затем тотчас же повторно гранулируется путем центрифугирования. Клеточный осадок снова суспензируется в 100 мл 30 мМ 2 и инкубируется во льду в течение 20 мин. Эти клетки снова собираются путем центрифугирования и снова суспензируются в 10 мл 30 мМ 2. Аликвота этих клеток, составляющая пол-мл, вводится в сшитую ДНК, приготовленную как в примере 29 А 2, приготавливается ДНК концентрацией 30 ммоль в 2. Смесь клетки - ДНК инкубируется во льду в течение 1 ч, нагревается при 42 С в течение 90 с и затем охлаждается на льду в течение 2 мин. Смесь клетки ДНК разбавляется в 10 мл средыи 125-миллилитровых колбах и инкубируется при 37 С в течение 1 ч. Аликвоты в количестве 100 мкл высеваются в чашках с -агаром, содержащих ампициллин, и инкубируются при 37 С до тех пор, пока не появляются колонии. Эти колонии индивидуально культивируются, и ДНК плазмиды отдельных колоний подвергаются анализу на ограничительный фермент и гель-электрофорезу. Выделение ДНК плазмиды осуществляется в небольших количествах согласно процедуре примера 29 А 1, но этап градиентане осуществляется до тех пор, пока интифицируются .К 12 МОрКС 283 РХ трансформанты. Ограничительная точка и функциональная карта плазмиды рКС 283 РХ представлены на фиг. 3. 4. Построение .К 12 МО/283-. 10 мкг ДНК плазмиды рКС 283 РХ, полученной согласно процедуре примера 29 А 1, растворяются в 20 мкл 10 Х высокосолевого буферного раствора, 20 мкл 1 мг/мл , 5 мкл ( 50 ед.) ограничительного фермента , 5 мкл ( 50 ед.) ограничительного ферментаи 150 мкл воды, и полученная реакционная смесь инкубируется при 37 С в течение 2 ч. Реакция прекращается и после осаждения вываренной сДНК эта ДНК снова суспензируется в 5 мкл буферного раствора ТЕ. Синтезируется и киназируется связывающее звено ДНК с однониточными концами ДНК, характерными для расщепления ограничительного ферментаи . Это связующее звено киназируется согласно процедуре примера 29 А 2. Связывающее звено ДНК имеет структуру, приведенную на фиг. 23. Приведенное связующее звено синтезируется из однониточных деоксилигонуклеотидов согласно уже хорошо известной в данной области процедуре. Эти однониточные деоксилигонуклеотиды могут быть синтезированы посредством выпускаемых промышленностью инструментов, таких как синтезатор ДНК 380 А,выпускаемых в продажу фирмой 1850,( 94404), который используется в химии фосфорамидитов. Известны также и другие процедуры синтеза ДНК. Обычный модифицированный метод фосфотриэфира для синтеза однониточной ДНК описывается в работеи др.,1977, , 1981056 ии др., 1978, . . . . , 755765. Кроме того, особенно предпочтительный способ синтеза ДНК описывается в работеи др., 1983, 11, 3227 и в работеи др., 1980,, 6890. Это связующее звено и вываренная с-плазмида рКС 283 РХ сшиваются в основном согласно процедуре примера 29 А 2. Сшитая ДНК образует желаемую плазмиду 283-. Ограничительная точка и функциональная карта плазмиды 283- представлены на фиг. 4. ДНК плазмиды 283- используется для трансформации .К 12 МО , и полученные трансформанты .Е 12 МО /283- идентифицируются в основном согласно процедуре примера 29 3. 5. Построение .К 12 МО /283-. Примерно 10 мкг ДНК плазмиды 283-, полученной согласно процедуре примера 29 А 1, растворяются в 20 мкл 10 Х высокосолевого буферного раствора, 20 мкл 1 мг/мл , 5 мкл ( 50 ед.) ограничительного ферментаи 155 мкл 2, и полученная реакционная смесь инкубируется при 37 С в течение 2 ч. Вываренная сДНК плазмиды 283- затем осаждается из реакционной смеси путем ввода трех объемов 95 -ного этанола и 1/10 объема 3 М ацетата натрия, инкубации в бане сухой лед - этанол в течение 5 мин и центрифугирования. Полученный осадок ДНК промывается 70 -ным этанолом, высушивается и повторно суспензируется в 2 мкл 10 Х Ник-трансляционного буферного раствора (0,5 моль Трис-, рН 7,2,16 3225 1 0,1 моль 4 и 1 ммоль ДТТ), 1 мкл раствора 2 мМ в каждом из деоксинуклеотидотрифосфатов, 15 мкл 2, 1 мкл ( 6 ед., как определено - ) фрагмента Кленова, который является большим фрагментом 1 ДНК полимеразы . , и 1 мкл 1 мг/мл . Полученная реакционная смесь инкубируется при 25 С в течение 30 мин, реакция прекращается в результате инкубирования раствора при 70 С в течение 5 мин. Связывающие звенья(5/3) киназируются и сшиваются с вываренной с ,обработанной фрагментом Кленова ДНК плазмиды 283- в основном согласно процедуре примера 29 А 2. После реакции сшивки ДНК вываривается примерно с 100 ед. Ва А в течение 2 ч при 37 С в высокосолевом буферном растворе. После вываривания сДНК подготавливается для сшивки согласно процедуре примера 29 А 2. Ограничительный фрагмент 5,9 кб Вазамыкается в кольцо путем сшивки и трансформируется в .К 12 МОсогласно процедуре примера 29 А 2 и 29 3. Трансформанты К 12 МО /283 идентифицируются, и затем получается плазмида 283- согласно процедуре примера 29 . Ограничительная точка и функциональная карта плазмиды 283- представлены на фиг. 5. 6. Построение .К 12 МО /32. Примерно 10 мкг плазмиды рКС 283 РХ вываривается с ограничительным фрагментомв высокосолевом буферном растворе, обрабатывается фрагментом Кленова и сшивается со связывающими звеньями(53) в основном согласно процедуре примера 29 А 5, с той разницей, что используются исходная плазмида, ограничительные фрагменты и связывающие звенья. После вываривания с ограничительным фрагментом, который образует в результате фрагмент 2,1 от ДНК, ограничительный фрагмент 4 кбзамыкает кольцо путем сшивки с получением плазмиды 283. Эта сшитая ДНК используется для трансформации .К 12 МОв основном согласно процедуре примера 29 3. После идентификации трансформант .К 12 МО /283 ДНК плазмиды 283 обрабатывается согласно процедуре примера 29 . Ограничительная точка и функциональная карта плазмиды 283 представлены на фиг. 6. Примерно 10 мкл плазмиды 283 вываривается в 200 мкл высокосолевого буферного раствора примерно с 50 ед. каждого из ограничительных фрагментови . После инкубирования реакционной смеси при 37 С в течение примерно 2 ч реакционная смесь подвергается электрофорезу на 0,6 гельагарозе с низкой температурой гелеобразования (фирма Отделение, ,04841) в течение 2 - 3 ч при 130 В и 75 мА в Трис-ацетатном буферном растворе. Этот гель окрашивается в разбавленном растворе этилиденбромида, и слой ДНК, составляющий ограничительный фрагмент 0,85 кб- , который визуально обнаруживается в длинноволновых ультрафиолетовых лучах, отсекается от данного геля в небольшом сегменте. Объем этого сегмента определяется по весу и плотности, и в пробирку, содержащую данный сегмент, вводится равный объем 10 ммоль Трис-, рН 7,6. Затем этот сегмент расплавляется путем инкубации при 72 С. Получается примерно 1 мкг ограничительного фрагмента 0,85 кб-плазмиды 283 в объеме примерно 100 мкл. Аналогичным образом плазмида 283- вываривается с ограничительными ферментамии , полученный ограничительный фрагмент 3 кб изолируется путем электрофореза на агарозном геле и обрабатывается для подготовки к сшивке. Ограничительный фрагмент 0,85 кб-плазмиды 283 сшивается с ограничительным фрагментом 0,3 кб-плазмиды 283- согласно процедуре примера 29 А 2. Сшитая ДНК составляет желаемую плазмиду 32. Ограничительная точка и функциональная карта плазмиды 32 представлены на фиг. 7. Плазмида 32 трансформируется в клетки .К 12 МОв основном согласно процедуре примера 29 3. ДНК плазмиды 32 приготавливается из трансформанта .К 12 МО/32 согласно процедуре, описанной в примере 29 . Анализ ДНК плазмиды 32 продемонстрировал,что более чем одно связующее звеноприкрепляется к обработанным фрагментом Кленова концамплазмиды рКС 283 РХ. Присутствие более чем одного связывающего звенане влияет на полезность плазмиды 32 или производных плазмиды 32 и может быть определено по присутствию ограничительной точки , которая образуется независимо от того, когда сшиваются оба связующих звенавместе друг с другом. Как возможный вариант, плазмида 32 может быть построена путем эксцизии фрагмента -и сшивки согласно первому разделу данного примера с описанием плазмиды 283-. 7. Построение .К 12 МО /47.. К 12 308/789 может быть получена из Северной региональной научно-исследовательской лаборатории в лиофилизированной форме под номеромВ-18216. Ограничительная точка и функциональная карта 789 представлены на фиг. 8. Плазмидная ДНК экстрагируется из данной культуры, согласно описанию примера 1, с той разницей, что температура инкубирования составляет 37 С. 10 мкг 789 суспензируются в 200 мкл буферного раствора(50 ммоль Трис-, рН 7,5, 60 ммольи 6 ммоль 2). Вводится 1 ед. , и реакционная смесь инкубируется в течение 5 мин при 37 С. Данный фермент инактивируется путем нагрева в течение 10 мин при 65 С. Затем вводятся 30 мкл 10 Х буферного 17 3225 1 раствора(200 ммоль Трис- (рН 8), 1 мольи 70 ммоль 2), 70 мкл 2 и 10 ед. , и реакционная смесь инкубируется в течение 1 ч. при 37 С. После этого вводятся 5 ед. щелочной фосфотазы и осуществляется инкубирование в течение 1 ч при 65 С. Фрагменты ДНК отделяются на 1-ном агарозном геле, и осуществляется очистка фрагмента ДНК (фиг. 9) размером в единичный фрагментный отрезок. Связывающее звено ДНК с отсеченным концом и с Ва -концом синтезируется в основном согласно процедуре примера 29 А 4. Это связывающее звено (показано на фиг. 9) имеет структуру, представленную на фиг. 24. Это связывающее звено киназируется и сшивается в вываренную Ва-плазмиду 789 в основном согласно процедуре примера 29 А 2. Эта смесь сшивки используется как трансформация клеток Е. Со К 12 308, и выделение плазмиды осуществляется с использованием данных трансформант согласно описанию примера 29 3. Выбираются некоторые плазмиды, которые содержат фрагмент(494 ) подходящего размера и фрагмент- Ва(628 ). Последовательность по меньшей мере двух из них определяется из последовательности от точки Вак уникальной точке , и выбирается один клон с желаемой последовательностью. Эта промежуточная плазмида обозначается как плазмида 120. Схематическое изображение данной процедуры, ограничительная точка и функциональная карта плазмиды 120 представлены на фиг. 9. Для выделения кодирующей - ДНК примерно 10 мкг плазмиды 120 вываривается в 200 мкл высокосолевого буферного раствора, содержащего примерно 50 ед. каждого из ограничительных ферментови Ва . Продукты вываривания отделяются посредством электрофореза на агарозном геле, и ограничительный фермент 0,6 кб - , который кодирует -, выделяется и препарируется для сшивки согласно процедуре, описанной в примере 29 А 6. Плазмида 32 также вываривается с ограничительными ферментамии , и ограничительный фрагмент 3,9 кб извлекается и приготавливается для сшивки. Ограничительный фрагмент 3,9 кбплазмиды 32 сшивается с ограничительным фрагментом 0,6 кб -плазмиды 120 в основном согласно процедуре примера 29 А 2, и в результате получается плазмида 47. Ограничительная точка и функциональная карта плазмиды 47 представлены на фиг. 10. Плазмида 47 трансформируется в .К 12 МОв основном согласно процедуре примера 29 3, и идентифицируются трансформанты .К 12 МО /47. ДНК плазмиды 47 получается от данных трансформант в основном согласно процедурам примера 29 А 1. 8. Построение .К 12 Р 308/121. Плазмида 12 заключает в себе чувствительный к температуре подавляющий- ген с 1857, и плазмида 322 заключает ген, придающий стойкость к тетрациклину. Плазмида 12 описана и заявлена в патенте США 4436815, выпущенном 13 марта 1984 года. Ограничительная точка и функциональная карта плазмиды 12 представлены на фиг. 11. Примерно 10 мкг плазмиды 12 вывариваются примерно с 50 ед. ограничительного ферментав 200 мкл высокосолевого буферного раствора при 37 С в течение 2 ч. Вываренная сДНК плазмиды 12 осаждается и обрабатывается фрагментом Кленова согласно процедуре примера 29 А 5. После протекания реакции Кленова вываренная, обработанная фрагментом Кленова ДНК плазмиды 12, снова замыкается в кольцо путем сшивки согласно процедуре примера 29 А 2. Эта сшитая ДНК, которая образует желаемую плазмиду 121, используется для трансформации .К 12 308 в основном согласно процедуре примера 29 3, с той разницей, что этот выбор основан на стойкости к тетрациклину (5 мкг/мл), а не к ампициллину. .К 12 308 имеет официальный номерВ-15624. После идентификации трансформант .К 12 308/121 из данных трансформант получается ДНК плазмиды 121 согласно процедуре примера 29 11. Примерно 10 мкг плазмиды 121 вывариваются примерно с 50 ед. ограничительного ферментав 200 мкл среднесолевого буферного раствора при 37 С в течение 2 ч. Вываренная сДНК плазмиды 121 осаждается и обрабатывается фрагментом Кленова согласно процедуре примера 29 А 5. После реакции с фрагментом Кленова вываренная с , обработанная фрагментом Кленова ДНК плазмиды 121, сшивается со связывающими звеньями Есо(53) в основном согласно процедуре примера 29 А 2. После сшивки связывающим звеном ДНК осаждается и затем повторно суспензируется примерно в 200 мкл высокосолевого буферного раствора, содержащего примерно 50 ед. ограничительного фермента Есо . Полученная реакционная смесь инкубируется при 37 С в течение примерно 2 ч. После вываривания с Есореакционная смесь помещается на агарозный гель, и ограничительный фрагмент 5,1 кб Есоочищается в основном согласно процедуре примера 29 А 6. Ограничительный фрагмент 5,1 кб Есоснова замыкается в кольцо путем сшивки согласно процедуре примера 29 А 2. Эта сшитая ДНК образует желаемую плазмиду 121. Эта ДНК плазмиды 121 трансформируется в .К 12 308 согласно процедуре пример 29 3, с той разницей, что выбор основывается на стойкости к тетрациклину, а не к ампициллину. 3225 1 После идентификации трансформант .К 12 308/121 получается ДНК плазмиды 2 в основном согласно процедуре примера 29 А 1. Ограничительная точка и функциональная карта плазмиды 121 представлены на фиг. 12. 9. Построение .К 12 308/110. Примерно 10 мг ДНК плазмиды 121 суспензируются в примерно 200 мл высокосолевого буферного раствора, содержащего примерно 50 ед. каждого из ограничительных ферментови Есо , и реакционная смесь вываривания инкубируется при 37 С в течение примерно 2 ч. Затем эта реакционная смесь помещается на агарозный гель, и ограничительный фрагмент 2,9 кб- Есоплазмиды 121 выделяется и подготавливается для сшивки в основном согласно процедуре примера 29 А 6. Примерно 10 мкг плазмиды 47 вывариваются с ограничительными ферментамиив 200 мкл высокосолевого буферного раствора при 37 С в течение 2 ч. Вываренная с-ДНК помещается на агарозный гель, и выделяется ограничительный фрагмент 2,7 кб - , который является источником репликации с частью стойкого к ампициллину гена и который подготавливается для сшивки согласно процедуре примере 29 А 6. В реакции разделения примерно 10 мкг ДНК плазмиды 47 вывариваются с ограничительными фрагментами Есои Вав 200 мкл высокосолевого буферного раствора при 37 С в течение 2 ч, и выделяется ограничительный фрагмент 1,03 кб- Ва , который заключает в себя новую транскрипционную и трансляционную активирующую последовательность и кодирующую - ДНК, и этот фрагмент подготавливается для сшивки согласно процедуре примера 29 А 6. Примерно 2 мкг полученного ограничительного фрагмента 1,03 кб Есо-используются для построения плазмиды 110. Ограничительные фрагменты 2,7 кб -и 1,03 кб Есо-плазмиды 47 сшиваются с ограничительным фрагментом 2,9 кб- Есоплазмиды 121 для построения плазмиды 110, и сшитая ДНК используется для трансформации .К 12 308 в основном согласно процедуре примеров 29 А 2 и 29 3, с той разницей, что основой для выбора трансформант является стойкость к тетрациклину, а не к ампициллину. В кодирующей зоне - присутствуют две распознавательные точки ограничительного фрагмента , которые не отражаются на ограничительной точке и на функциональных картах на фигурах данной заявки. Ограничительная точка и функциональная карта плазмиды 110 представлены на фиг. 13. 10. Построение .К 12 308/110. а. Построение .К 12 308/110. Примерно 1 мкг ДНК плазмиды 110 вываривается с ограничительным ферментомв 20 мкл общего объема, содержащего 2 мкл 10 Х высокосолевого буферного раствора (1 моль , 0,50 моль Трис-,рН 7,5, 0,10 моль 2 и 10 ммоль дитиотреитола) и 3 ед. фермента , в течение 1 ч при 37 С. Реакционная смесь экстрагируется фенолом/хлороформом, и ДНК осаждается этанолом. Вываренная сДНК плазмиды 110 растворяется в 50 мкл 1 буферного раствора Кленова (40 ммоль К 2 НР 4, рН 7,5, 6,6 ммоль 2, 1 ммоль 2-меркаптоэтанола, 33 мкмоль , 33 мкмольи 33 мкмоль ТТР). 2 мкл ( 10 ед.,) крупных фрагментов ДНК полимеразы 1 Е. , известных как фрагменты Кленова, вводятся в данный раствор и смешиваются с ДНК, и полученная реакционная смесь инкубируется при 16 С в течение 1 ч. Реакция завершается путем экстрактации фенолом, и ДНК очищается обычными приемами. Вываренная с , обработанная фрагментом Кленова ДНК затем сшивается с 4 ДНК лигазой при 4 С в течение 16 ч. Полученная ДНК используется для обычной трансформации штамма Е.К 12 308( В-15624). Трансформанты отбираются на чашках с -агаром, содержащих 100 мкг/мл ампициллина,и плазмиды извлекаются из стойких колоний путем быстрой щелочной экстракции, как описано в работеи . Отбирается плазмида (110 на фиг. 14) с отсутствием точки .. Построение фага 110 В путем точечно-специфического мутагенеза. Осуществление процедуры удаления точкив гене, сообщающем стойкость к тетрациклину путем точечно-специфического мутагенеза, представлено с правой стороны фиг. 14.. Построение фага 3 Тс 3. Плазмида 110 служит в качестве источника, придающего стойкость к тетрациклину гена. Примерно 50 мкг плазмиды 110 в 50 мкл буферного раствора ТЕ вводятся в 25 мкл буферного раствора 10 Хи 170 мкл Н 2. Примерно 5 мкл ( 50 ед.) ограничительного ферментавводятся в раствор ДНК плазмиды 110, и полученная реакционная смесь инкубируется при 37 С в течение 2 ч. Примерно 13 мкл 2 М Трис-, рН 7,4, и 5 мкл ( 50 ед.) ограничительного ферментавводятся в вываренную сДНК плазмиды 110, и реакционная смесь инкубируется в течение более 2 ч при 37 С. Реакция прекращается в результате экстракции реакционной смеси насыщенным ТЕ-фенолом, фенол удаляется путем экстракций хлороформом. Вываренная с- ДНК плазмиды 110 затем извлекается путем осаждения и центрифугирования, помещается на 1 -ный агарозный гель, и крупный ограничительный фермент 4,3 кб- выделяется и очищается. Примерно 5 мкг фага 13 8 растворяются в 50 мкл буферного раствора ТЕ, и затем выпариваютсяи, как описано выше. 3225 1 Отсеченная-ДНК фага 13 8 очищается, как описано для 110, с той разницей, что осуществляются выделение и очистка ограничительного фрагмента 7,25 кб. Примерно 100 нг фрагмента 4,3 кб-празмиды 110 смешиваются с примерно 100 нг фрагмента 7,25 кб-фага 3 тр 18, 2 мкл 10 Х буферного раствора лигазы, 1 мкл ( 100 ед.) Т 4 ДНК лигазы и 14 мкл Н 2 О. Реакционная смесь сшивки инкубируется при 15 С в течение 1,5 ч, сшитая ДНК составляет ДНК желаемого фага 3 3. Ограничительная точка и функциональная карта фага 3 3 представлены на фиг. 14. 1 мл ночной культуры Е.К 12 109 (Е.К 12 101 получена из, может использоваться вместо Е.К 12 109) используется для инокулирования 50 мл питательного бульона(50 мл), и полученная культура инкубируется при 37 С с аэрацией до тех пор, пока 660 (оптическая плотность) не будет составлять от 0,3 до 0,4. Клетки повторно суспензируются в 25 мл и 10 ммоль , инкубируются на льду в течение 10 мин и извлекаются путем центрифугирования. Эти клетки помещаются в 1,25 мл 0,75 мМ 2, 200 мкл аликвота этих клеток удаляется, вводится в 10 мкл сшитой ДНК, полученной, как описано выше, и инкубируется на льду в течение 40 мин. Затем смесь клетки - ДНК инкубируется при 92 С в течение 2 мин, и различные аликвоты (1,10 и 100 мкл) удаляются и вводятся в 3 мл верхнего агара (бульон с 0,5 -ным агаром, поддерживаемым в расплавленном состоянии при 45 С), который содержит также 50 мкл 2-, 50 мкл 100 мМи 200 мкл Е.К 12 109 в фазе логарифмического роста. Затем верхняя клеточная часть агаровой смеси высевается в чашках с -агаром, содержащих 40 мкг/мл(5-бромо-4-хлоро-3-индолилтиогалактозид) и 0,1 ммоль(изопропилтиогалактозид), и чашки инкубируются в течение ночи при 37 С. На следующее утро несколько прозрачных, но не синих колоний используются для инокулирования 2 мл-буольна, и образующиеся в результате культуры инкубируются при 37 С с аэрацией в течение 2 ч. Отсутствие синего окрашивания показывает, что происходит желаемая вставка ДНК. Затем данные культуры центрифугируются, и 200 мкл полученного поверхностного слоя вводятся в 10 мл культур (5500,5) Е.К 12 109, выращенных при 37 С с аэрацией. Эти культуры инкубируются еще в течение 30 мин при 37 С,затем эти клетки выпадают в осадок в результате центрифугирования и используются для получения репликаторной формы рекомбинантного фага, который они содержат. Из этих клеток выделяется двухниточная репликаторная форма фаговой ДНК с использованием процедур (в меньших объемах), описанных в примере 1. Трансформанты, содержащие фаговую 13 Тс 3 ДНК, идентифицируются путем анализа на ограничительный фрагмент их фаговой ДНК.. Получение однониточной ДНК фага 13 Тс 3. 1,5 мл ночной культурыК 12 109/3 Тс 3 центрифугируются, и 100 мкл верхнего слоя, содержащего фаг 13 Тс 3, используются для инокуляции 25 мл культурыК 12 109 с оптической плотностью 660 примерно 0,4 - 0,5. Данная культура инкубируется примерно в течение 6 ч при 37 С с аэрацией, и затем культура центрифугируется, образованный верхний слой примерно 20 мл переносится в новую пробирку. Примерно 2 мл раствора, содержащего 20 полиэтиленгликоля (РЕС) 6000 и 14,6, вводятся в поверхностный слой, который затем инкубируется на льду в течение 20 мин. Поверхностный слой центрифугируется в течение 25 мин со скоростью 7000 об/мин, и образующийся осадок, который содержит однониточную ДНК фага 13 Тс 3, снова суспендируется в 500 мкл буферного раствора ТЕ. Раствор ДНК двукратно экстрагируется насыщенным ТЕ-фенолом и двукратно - хлороформом. Затем однониточная ДНК осаждается с использованиеми этанола и центрифугируется. Полученный осадок промывается 70 -ным этанолом, высушивается и затем растворяется в 60 мкл 2.Мутагенез. Фрагмент однониточной ДНК, используемый в процессе мутагенеза, синтезируется в автоматическом синтезаторе ДНК. Этот фрагмент имеет последовательность 53 и является гомологом зоны, окружающей точку(53), в придающем стойкость к тетрациклину гене от плазмиды 322, с той разницей, что А-группа вторая от 5-конца (или третья от 3-конца), является С в плазмиде 322. Такое видоизменение не меняет аминокислотный состав белка, придающего стойкость к тетрациклину, но исключает точку. Примерно 10 пмоль мутагенной затравки и универсальной затравки М 13 (. .6009, Гатесбург,20760) обрабатываются по отдельности 10 ед.полинуклеотидокиназы Т 4 в 20 мкл буферного раствора киназы 1 (60 ммоль Трис-, рН 7,8, 15 ммоль 2-меркаптоэтанола, 10 ммоль 2 и 0,41 мкмоль АТР) в течение 30 мин при 37 С. Обработанные киназой ДНК используются в описанной ниже процедуре мутагенеза. Реакция ренатурации (анилирования) осуществляется путем смешивания друг с другом 300 нг (1,2 мкл) однониточного фага 13 Тс 3, 1 пмоль (2 мкл) универсальной затравки, 1 пмоль (2 мкл) мутагенной затравки, 2 мкл 10 Х ренатурирующего буферного раствора (100 ммоль Трис-, рН 7,5, 1 ммоль ЭДТА и 500 ммоль) и 12,8 мкл 2. Реакционная смесь инкубируется при 80 С в течение 2 мин при температуре 50 С в течение 5 мин, а затем охлаждается до комнатной температуры. 20 3225 1 Реакция удлинения цепи осуществляется путем ввода 5 мкл 10 Х буферного раствора вытягивания (или удлинения) (500 ммоль Трис-, рН 8, 1 ммоль ЭДТА и 120 ммоль 2), 5 мкл 2 мМ , 1 мкл раствора 6 мМ каждого из , ТТР и , 1 мкл ( 2 ед., , - , 800,08854) фермента Кленова, 1 мкл (100 ед.) Т 4 ДНК лигазы и 17 мкл 2 в смесь ренатурированной ДНК. Реакционная смесь удлинения цепи инкубируется при комнатной температуре в течение 1 ч,затем при 37 С в течение 2,5 ч и затем в течение ночи при 4 С. Реакция прекращается путем осуществления двух экстракций насыщенным ТЕ-фенолом, после которых осуществляются две экстракции СНС 13. ДНК осаждается этанолом и ацетатом натрия . ДНК извлекается путем центрифугирования и снова суспензируется в 50 мкл 2, и затем 6 мкл 10 Х буферного растворавводятся в раствор ДНК. Этот раствор ДНК распределяется поровну в трех пробирках. В две из этих пробирок вводится примерно 200 ед. ( ) нуклеазы 1. Одна реакционная смесьинкубируется при комнатной температуре в течение 5 мин, другая в течение 10 мин. Реакции прекращаются в результате экстракции реакционной смеси два раза насыщенным ТЕ-фенолом. После фенольных экстракций осуществляются две экстракции хлороформом, затем из данной реакционной смеси осаждается ДНК ацетатом натрияи этанолом. Необработанный образец ДНК служит в качестве отрицательного контрольного образца. Обработанные образцы сохраняются изолированными друг от друга в ходе последующей процедуры, но результаты получаются одинаковыми. Выпавшие осадки ДНК повторно суспензируются в 20 мкл Н 2, и используется 10 мкл полученного раствора для трансформации Е.К 12 109 (Е.К 12 101 также может быть использован) согласно процедуре, осуществляемой для построения фага 13 3, с той разницей, что в чашки не вводится ни ,ни -. Двухниточная репликаторная форма ДНК от примерно 48 колоний выделяется, как описано выше,и отбирается на присутствие ограничительной точки Ва . Изоляты без точки Вадополнительно отбираются с получением однониточной ДНК, как описано выше. Аминокислотная последовательность однониточной ДНК определяется с использованием определения дидеокси последовательности (. . , Методы в энзимологии, 65560 - 580). Желаемый изолят обозначается как 110 (фиг.14). с. Построение плазмиды 110. Примерно 50 мкг репликаторной формы фага 110 ДНК вывариваются в 250 мкг 1 буферного раствора(50 ммоль , 6 ммоль Трис-, рН 7,5, 6 ммоль 2 и 6 ммоль -меркаптоэтанола), содержащего 50 ед. ограничительного фермента , при 37 С в течение 2 ч. 5 мкл 5 Мзатем вводятся в вываренную сфаговую 110 ДНК с последующим вводом 5 мкл ( 50 ед.) ограничительного фермента . Вываривание продолжается в течение 2 ч при 37 С. Затем желаемый фрагмент 422 вр , содержащий мутированную зону гена, придающего стойкость к тетрациклину, выделяется из акриламидного геля согласно известным стандартным процедурам. ДНК плазмиды 110 вываривается сив идентичных условиях, с той разницей, что вместо плазмиды 110 используется плазмида 110. Ограничительный фрагмент 6,1 кб - плазмиды 110 очищается от агарозы. Построение желаемой плазмиды 110 осуществляется путем сшивки друг с другом по 100 нг каждого из- фрагментов 110 ( 6,1 кб) и 110 ( 422 вр) путем общеизвестных процедур. Ограничительная точка и функциональная карта плазмиды 110 представлены на фиг. 15. Желаемая плазмида 110 сообщает стойкость к тетрациклину для 10 мкг/мл тетрациклина в Е. , но в сообщающем стойкость к тетрациклину гене отсутствует точка. 11. Построение плазмиды . Плазмида 110 содержит единственную ограничительную точку , которая удаляется при протекании нижеследующих реакций. Примерно 1 мкг плазмиды 110 вываривается ссогласно процедуре примера 29 А 2, с той разницей, что используются ограничительный ферменти 10 Х буферный раствор(500 ммоль , 100 ммоль Трис-, рН 7,9 и 100 ммоль 2). Вываренная сДНК затем обрабатывается фрагментом Кленова согласно описанию примера 29 А 5, с той разницей, что вводится лишь, а не все четыре . Затем ДНК осаждается и повторно суспензируется в 50 мкл буферного раствора бобовой нуклеазы(50 ммоль ацетата натрия, рН 5, 30 ммольи 1 ммоль 4). 1 ед. бобовой нуклеазы(промышленно выпускается) вводится в эту суспензию, и реакционная смесь инкубируется при 30 С в течение 30 мин. Затем пробирка помещается в лед, ивводится в концентрации 0,2 моль, после чего смесь экстрагируется фенолом (хлороформом), осаждается этанолом и повторно суспензируется в 10 ммоль Трис- (рН 8). Затем ДНК подвергается самосшивке и трансформируется в клетки Е.согласно описанию примеров 29 3 и 29 А 4. Полученная плазмида представляет собой вываренную плазмиду 111. 12. Построение плазмиды 126. Примерно 26 мкг плазмиды 111 вывариваются суказанным ниже образом. Буферный раствор 10 Хсостоит из 600 ммоль Трис-, 100 ммоль 2, 1 мольи 10 ммоль 2-меркаптоэтанола, рН 21 3225 1 7,5 (при 37 С), 50 мкл буферного раствора 10 Х , 15 мкл(10 ед/мкл) и 185 мкл Н 2 вводятся в 250 мкл воды, содержащей примерно 25 мкл плазмиды 110. Вываривание осуществляется при 37 С в течение 1 ч. 110, вываренная с , затем экстрагируется в феноле, вводится 1/10 объема этанола, смесь инкубируется в бане сухой лед-этанол в течение 5 мин и затем центрифугируется. Осажденная ДНК повторно суспензируется в 50 мкл Н 2. Вываренная сплазмида 111, вываривается вследующим образом. 0,2 мкл Ва(10 ед/мкл), 10 мкл буферного раствора(100 ммоль Трис-, 50 ммоль 2,1 мольи 10 ммоль 2-меркаптоэтанола, рН 8, при 37 С) и 90 мкл Н 2 вводятся в 50 мкл 110, вываренного с , полученного, как указано выше. Вываривание осуществляется в течение 5 мин при 37 С. Вываренная 111 экстрагируется в феноле, вводится 1/10 объема 3, после чего вводятся 3 объема этанола. Осажденная ДНК снова суспензируется в 50 мкл 10 мМ Трис, 1 ммоль ЭДТА, рН 8, буферном растворе. 111, вываренная си, затем помещается на агарозный гель, и выделяется слой ДНК примерно 5,8 кб. Плазмида 126 получается путем сшивки фрагмента 5,8 кб 111 со связывающими звеньямиии синтетическим геном, кодирующим ЕК-бычий гормон роста, который содержит точкуу его 5-конца и точку Вау его 3-конца. Последовательностьдополучается с использованием метода построения стандартной олигонуклеотидной последовательности, и она заключает в себе последовательность, приведенную на фиг. 25. Указанная последовательность построена путем химического синтеза обеих нитей с последующим смешиванием с целью гибридизации. Кодирующий ген построен из 16 химически синтезированных сегментов однониточной ДНК, заключающих в себе от 71 до 83 нуклеотидов, которые совместно друг с другом составляют дополняющие нити всего гена. Синтез осуществляется в устройстве и включает последовательность, приведенную на фиг. 26 и 27. Построение плазмиды 126 осуществляется путем сшивки нижеследующих компонентов 0,28 мкг фрагмента 5,8 кб, полученного из плазмиды 110 после полного вываривания си частичного вываривания с Вав общем объеме 2 мкл,0,18 мкг синтетического гена, кодирующего производное фактора бычьего роста, который имеет конец 5, соответствующий точке , и конец 3, соответствующий точке Вав общем объеме 2,5 мкл, 8,75 пмоль химически синтезированного-связывающего звена в 1 мкл. Эти компоненты плазмиды .вводятся в 8 мкл 5 Х буферного раствора сшивки 250 ммоль Трис, 50 ммоль 2, 5 ммоль АТР, 5 ммоль ДТТ, 25 об./об. полиэтиленгликоля 8000, рН 7,6, 2 мкл лигазы и 16,5 мкл Н 2. Эта смесь сшивки инкубируется в течение ночи при 16 С. Далее замкнутая в кольцо плазмида 126 используется для трансформации клеток Е.308 согласно способу примера 29 3. Ограничительная точка и функциональная карта плазмиды 126 представлены на фиг.16. В. Построение выраженной плазмиды человеческого проинсулина 72. 1. Построение плазмиды 45. Ген человеческого проинсулина сначала синтезируется обычным образом и клонируется в плазмиду РС 18 (промышленно выпускаемая ). Этот ген синтезируется стандартными приемами и заключает в себе последовательность, представленную на фиг. 28. Один из клонов, имеющий правильную последовательность, выбирается для продуцирования очищенной от хлорида цезия ДНК. Данная плазмида извлекается путем стандартной процедуры (см., например, публикацию Молекулярное клонирование. Лабораторное пособие, 1982 г., .Т.. .,.,, Нью Йорк). Примерно 6 мкл (29 мкг) этой ДНК плазмиды вводятся в 20 мкл 10 Х буферного раствора(150 ммоль , 10 ммоль Трис-, рН 7,8, 6 ммоль 2, 6 ммоль 2-меркаптоэтанола, 100 мкг/мл ), 5 мкл ограничительного фермента( 40 ед.) и 169 мкл Н 2. После перемешивания реакционная смесь инкубируется при 37 С в течение 2 ч. ДНК осаждается путем приготовления 0,3 моль , ввода трех объемов этанола, перемешивания, охлаждения до 70 С и центрифугирования. Осажденная ДНК промывается 70 -ным этанолом (1 мл), высушивается и растворяется в 20 мкл 10 Х Вабуфера (150 ммоль , 6 ммоль Трис-, рН 7,9, 6 ммоль 2, 100 мкг/мл ), 2 мкл ограничительного фермента(40 ед.) и 178 мкл Н 2. После слабого перемешивания реакционная смесь инкубируется при 37 С в течение 2 ч. ДНК снова осаждается тремя объемами этанола, как определено выше, и подвергается электрофорезу на 1 -ном расплавленном агарозном геле (,морская агароза). Желаемый фрагмент ДНК, соответствующий примерно 270 вр, срезается с геля, и затем ДНК извлекается путем расплавления агарозы и пропускания через колонку - (,Ке . ), согласно процедуре, рекомендуемой продавцом. После осаждения и сушки ДНК выдерживается в 30 мкл 10 мМ Трис-, рН 8. Примерно 15 мкг плазмиды 126 (из примера 29 А 12) суспензируются в 20 мкл 10 Х буферного раствора , 5 мкл ограничительного фермента(40 ед.) и 2 (175 мкл), слабо перемешиваются, и смесь инкубируется при 37 С в течение 2 ч. После инкубации ДНК осаждается тремя объемами этанола, как и выше, высушивается и затем повторно суспензируется в 20 мкл 10 Х буферного раствора, 2 мкл ограничительного фермента(40 ед.) и 178 мкл воды. После слабого перемешивания реакционная смесь инкубируется при 37 С в течение 2 ч. ДНК снова осаждается тремя объемами этанола, и осуществляется 22 3225 1 электрофорез на 1 -ном низкоплавком агарозном геле. Наиболее крупный фрагмент, соответствующий векторной ДНК, срезается с этого геля, и ДНК извлекается путем осуществления процедуры в колонке -, как описано выше. После осаждения и сушки векторная ДНК выдерживается в 35 мкл 10 мМ Трис, рН 8. Примерно 2,5 мкл векторной ДНК смешиваются с 12 мкл генного фрагмента очищенного человеческого инсулина, 4 мкл 10 мМ АТР, 0,5 мкл и 1 моль дитиотреитола, 5 мкл 10 Х буферного раствора лигазы (50 ммоль Трис-, рН 7,6, и 100 ммоль 2), 26 мкл воды и 0,5 мкл 4 ДНК лигазы (, 3,5 ед.). Эта реакционная смесь инкубируется при 4 С в течение 16 ч. Сшитая смесь разбавляется 50 мкл 10 мМ Трис (рН 7,6) и 3 мкл 1 М 2 и затем последовательно трансформируется в Е.К 12 308 согласно описанию примера 29 3. Клетки высеваются на чашках ТУ, содержащих 5 мкг/мл тетрациклина, и затем инкубируются в течение ночи при 32 С. Плазмиды от 3 мл культур выделяются из стойких к тетрациклину колоний путем быстрой щелочной экстракции, как описано в публикации Молекулярное клонирование. Лабораторное пособие, 1982 г., ред.Т.,. . и., Нью Йорк, с. 368-369. Присутствие нужного генного фрагмента человеческого проинсулина определяется путем процедуры миниотбора с использованием электрофореза на полиакриламидном геле для анализа вываренного с /фрагмента. Плазмиды с правильными по размеру вставками (примерно 316 вр) отбираются для амплификации и очистки. Плазмида, содержащая ген человеческого проинсулина, представляет собой 145. Ограничительная точка и функциональная карта плазмиды 145 представлены на фиг. 18. 2. Построение плазмиды 164. Примерно 30 мкг плазмиды 145 суспензируются в 20 мкл 10 Х буферного раствора , 5 мкл ограничительного фермента(, 40 ед.) и 175 мкл Н 2 с осторожным перемешиванием, и смесь инкубируется при 37 С в течение 1 ч. Затем в реакционную смесь вводится 2 мкл ограничительного фермента(, 40 ед.), и осуществляется инкубирование при 37 С в течение еще 2 ч. ДНК осаждается тремя объемами этанола и 0,3 мольи подвергается электрофорезу на 1 -ном низкоплавком агарозном геле. Более мелкий ограничительный фрагмент (примерно 270 вр) / ,кодирующий ген человеческого проинсулина, срезается с геля, и ДНК извлекается путем прохождения через колонку -, как описано выше. После осаждения и сушки ДНК выдерживается в 30 мкл 10 мМ Трис,рН 8. Затем к этой ДНК (30 мкл) добавляется 20 мкл 10 Х буферного раствора(50 ммоль , 6 ммоль Трис-, рН 8, 10 ммоль 2, 6 ммоль 2-меркаптоэтанола, 100 мкг/мл ), 5 мкл ограничительного фермента(, 20 ед.) и 175 мкл 2. После осторожного перемешивания данная реакционная смесь инкубируется при 37 С в течение 2 ч. ДНК осаждается тремя объемами этанола и 3 ммоль(20 мкл) и затем подвергается электрофорезу на 1,2 -ном низкоплавком агарозном геле. Более крупный ограничительный фрагмент /(примерно 156 вр) срезается с геля, и затем ДНК извлекается путем прохождения через колонку -, как описано выше. После осаждения и сушки ДНК выдерживается в 30 мкл 10 мМ Трис, рН 8. ДНК ( 115 вр), соответствующая ограничительному фрагменту / гена человеческого проинсулина, получается путем синтеза. Первый этап заключается в синтезе четырех однониточных деоксирибоолигонуклеотидов посредством синтезатора ДНКмодели 380 В. Нуклеотидные последовательности этих четырех олигонуклеотидов представлены на фиг. 29. После очистки каждого олигонуклеотида путем электрофореза на полиакриламидном геле олигонуклеотиды два и три фосфорилируются согласно описанию публикаций. ... . и. . (1979 г.), Методы в энзимологии. Ред..,Ресс, Нью Йорк, 68, 109-151. Фосфорилированные олигонуклеотиды два и три ( 715 пмоль каждого) затем смешиваются с олигонуклеотидами один и два ( 860 пмоль), ренатурируются и сшиваются в буферном растворе (200 мкл), содержащем 50 ммоль Трис- (рН 7,6), 10 ммоль 2 , 10 ммоль ДТТ, 59 мкмоль АТР и 20 ед. Т 4 ДНК лигазыв течение 16 ч при 4 С. Продукт сшивки очищается на 15 -ном полиакриламидном геле. ДНК извлекается из гелевого слоя путем электрофореза с последующим обессоливанием в колонкеС-50. Выход желаемого сшитого продукта составляет 485 пмоль. Примерно 100 пмоль этой ДНК фосфорилируется в буферном растворе (50 мкл), содержащем 50 ммоль Трис (рН 7,6), 10 ммоль 2, 10 ммоль ДТТ и АТР, как описано в публикации. . и др., 1979 г. Методы в энзимологии, 68, 109-151. После фильтрации через колонкуС-50 ДНК выдерживается в 50 мкл 10 мМ Трис, рН 8. Примерно 2,5 мкл векторной ДНК (вываренной в -126) смешиваются с 18 мкл ограничительного фрагмента /от плазмиды 145 и 10 мкл (10 пмоль) синтетического связывающего звена / в буферном растворе (50 мкл), содержащем 50 ммоль Трис (рН 7,6), 10 ммоль 2,10 ммоль ДТТ, 800 мкл АТР и 3,5 ед. Т 4 ДНК лигазы. Реакционная смесь инкубируется при 4 С в течение ночи и затем трансформируется в .К 12 308 согласно процедуре примера 29 3. Желаемая трансформанта .К 12 308/164 идентифицируется путем анализа ее плазмидной ДНК. Нужная 23 3225 1 трансформанта выращивается при 30 С в среде Т 4, содержащей 5 мгк/мл тетрациклина, до достижения 550 примерно 0,2 (ранняя логарифмическая фаза роста) и затем при 42 С в течение 3-3,5 ч для индуцирования синтеза человеческого проинсулина. Клетки выпадают в осадок и лизируются путем добавления буфера (0,125 моль Трис -НС 1, рН 6,8, 2, 30 глицерина, 1 моль 2-меркаптоэтанола, 6 моль мочевины). Образцы нагреваются до 97 С в течение 2 мин до помещения их на акриламидный гель. Слои легко визуально просматриваются путем окрашивания красителем Кумаси Брильянтовым синим. Для количественного определения процента клеточного белка используется сканирующий гелевый денситометр. Ограничительная точка и функциональная карта плазмиды 164 А представлены на фиг. 19. 3. Построение плазмиды 172. Примерно 25 мкг плазмиды 145 суспензируются в 15 мкл 10 Х буферного раствора(200 ммоль( , 27 ед.) и 129 мкл Н 2 О, осторожно перемешиваются и инкубируются при 37 С в течение 6 ч. После инкубации ДНК осаждается, высушивается и повторно суспензируется в 10 мкл 10 Х буфера(150 ммоль , 6 ммоль Трис-, рН 7,9, 6 ммоль 2, 7 ммоль 2-меркаптоэтанола, 100 мкг/мл), 3 мкл ограничительного фермента( , 36 ед.) и 77 мкл 2. Реакционная смесь осторожно перемешивается и инкубируется при 37 в течение 4 ч. ДНК снова осаждается тремя объемами этанола и ацетата натрия, подвергается электрофорезу на 1 -ном низкоплавком агарозном геле. Нижний слой, соответствующий примерно 85 вр ограничительного фермента / гена человеческого проинсулина, срезается с геля, и ДНК извлекается путем осуществления процедуры в колонке -. После осаждения и сушки ДНК выдерживается в 30 мкл 10 мМ Трис, рН 8. Примерно 15 мкг плазмиды 164 отсекаются с ограничительным ферментомисогласно описанной выше процедуре. Верхний слой, соответствующий векторному фрагменту /, выделяется из агарозного геля путем осуществления процедуры в колонке -. После осаждения и сушки ДНК выдерживается в 30 мкл 10 мМ Трис, рН 8. Примерно 5 мкл векторной ДНК 164 А, вываренной с /, смешиваются с 5 мкл ограничительного фрагмента / от плазмиды 145 с буферном растворе (50 мкл), содержащем 50 ммоль Трис-, рН 7,6, 10 ммоль 2 , 10 ммоль ДТТ, 800 ммоль АТР и 6 ед. Т 4 ДНК лигазы. Реакционная смесь инкубируется при 4 С в течение ночи и используется для трансформации клеток .К 12 308,которые превращаются в компетентные клетки путем стандартной обработки 2. Трансформанты выбираются на ТУ агаровых чашках, содержащих 5 мкг/мл тетрациклина. Плазмиды выделяются из стойких к тетрациклину колоний и анализируются путем вываривания с ограничительными нуклеазамии . Выстраивается аминокислотная последовательность ДНК от положительных клонов с использованием системы построения последовательности (. . ). Плазмиды с желаемой правильной последовательностью отбираются для амплификации и очистки. Таким образом изолируется трансформанта .К 12 308/172. Выражение и аккумуляция человеческого проинсулина, заключающего в себе данную плазмиду, анализируются путем визуализации всего клеточного белка с последующим электрофоретическим отделением в 15 -ной полиакриламидной матрице. Ограничительная точка и функциональная карта плазмиды 172 представлены на фиг. 20. С. Построение 308/173 и 174. Примерно 20 мкг плазмиды 172 или 145 суспензируются в 20 мкл 10 Х буфера(150 ммоль, 6 ммоль Трис-, рН 7,9, 6 ммоль 2, 7 ммоль 2-меркаптоэтанола, 100 мкг/мл ), вводятся 2 мкл ограничительного фермента( , 24 ед.), смесь осторожно перемешивается и инкубируется при 37 С в течение 4 ч. После инкубирования ДНК осаждается, высушивается и снова суспензируется в 10 мкл 10 Х буферного раствора(100 ммоль , 10 ммоль Трис-, рН 8, 5 ммоль 2,1 ммоль 2-меркаптоэтанола, 100 мкг/мл ), 5 мкл ограничительного фермента( , 40 ед.) и 75 мкл Н 2 О. Реакционная смесь осторожно перемешивается и инкубируется при 37 С в течение 4 ч. ДНК снова осаждается этанолом и ацетатом натрия и подвергается электрофорезу на 1 -ном низкоплавком агарозном геле. Ограничительный фермент /Вапримерно 320 вр, соответствующий гену человеческого проинсулина, извлекается из геля путем осуществления процедуры в колонке -. После осаждения и сушки ДНК снова суспензируется в 20 мкл 10 Х буферного раствора Х (25 ммоль , 10 ммоль Трис-, рН 7,5, 10 ммоль 2, 10 ммоль 2-меркаптоэтанола, 10 мкг/мл ), 10 мкл ограничительного фермента(, 10 ед.) и 170 мкл воды. Реакционная смесь осторожно перемешивается и инкубируется при 37 С в течение 4 ч. После инкубирования ДНК осаждается этанолом и ацетатом натрия и подвергается электрофорезу на 1 -ном низкоплавком агарозном геле. Более крупная ДНК,соответствующая ограничительному фрагменту /Х примерно 200 вр, срезается с геля, и эта ДНК выделяется путем осуществления процедуры в колонке -, как описано выше. Эта ДНК выдерживается в 30 мкл 10 мМ Трис, рН 8. ДНК на 51 вр, соответствующая ограничительному фрагменту Х/Вагена человеческого проинсулина, выравнивается в последовательность, которая определена ниже. Два однониточных деоксирибоолигонуклеотида, приведенных на фиг. 30, синтезируются с помощью синтезатора ДНК,(мо 24 3225 1 дели 380 В), и очищаются путем электрофореза на полиакриламидном геле в присутствии 7 моль мочевины. После изоляции оба нуклеотида фосфорилируются согласно процедуре в описании. .,.. . и. . (1979 г.). Методы энзимологии. Ред.., . ., 68, 109-151, и ренатурируются с образованием желаемого связывающего звена. Примерно 19 мкг плазмиды 126 (из примера 29 А 12) сусупензируются в 20 мкл 10 Х буферного раствора(150 ммоль , 6 ммоль Трис-, рН 7,9, 6 ммоль 2, 7 ммоль 2-меркаптоэтанола, 100 мкг/мл ), 2 мкг органического фермента( , 24 ед.) и 178 мкл воды. Реакционная смесь осторожно перемешивается и инкубируется при 37 С в течение 2 ч. После этого инкубирования вводятся 4 мкл 5 Ми 2 мкл ограничительного фермента(, 20 ед.), и инкубирование продолжается еще в течение 2 ч при 37 С. Затем ДНК осаждается эталоном и ацетатом натрия стандартным приемом и подвергается электрофорезу на 1 -ном низкоплавком агарозном геле. Верхний слой, соответствующий векторной ДНК / , выделяется путем осуществления процедуры в колонке -. После осаждения и сушки ДНК выдерживается в 30 мкл 10 мМ Трис, рН 8. Примерно 5 мкл векторной ДНК 126, вываренной с / , смешиваются с 10 мкл ограничительного фрагмента / от 145 или 172 и 4 мкл киназированного связующего /Ва , приготовленного, как указано выше, в буферном растворе (50 мкл), содержащем 50 мкмоль Трис (рН 7,6), 10 ммоль 2, 10 ммоль ДТТ, 800 мкмоль АТР и 6 ед. Т 4 ДНК лигазы. Реакционная смесь инкубируется при 4 С в течение ночи и используется для трансформации штамма .К 12 308 согласно процедуре примера 29 3. Желаемые трансформанты .К 12 308/173 (если они образованы от векторного фрагмента 172) и .К 12 308/174 (если они образованы от векторного фрагмента 145) идентифицируются путем анализа их плазмидной ДНК и белка до и после индукции при 42 С. Ограничительная точка и функциональная карта плазмиды 173 представлены на фиг. 21. Примерно 200 нг ДНК плазмиды от мини-препаративной обработки трансформируются в клетки .201, и трансформанты отбираются на чашках с 2 Х ТУ-агаром, содержащих 15 мкг/мл тетрациклина. Белки,выраженные этими трансформантами, анализируются, и желаемые трансформанты подтверждаются по продуцированию человеческого инсулина. Таким образом, выделяется .201 173 и . 201/174.. ПостроениеК 12 308/175, 176, 177 и 178. Примерно 12 мкг плазмиды 172 суспензируются в 15 мкл 10 Х буфера(100 ммоль , 50 ммоль Трис-, рН 7,5, 10 ммоль 2, 1 ммоль ДТТ, 100 мкг/мл ), 2,5 мкл ограничительного фермента(20 ед.) и 133 мкл воды. Реакционная смесь осторожно перемешивается и инкубируется при 37 С в течение ночи. После инкубирования ДНК осаждается этанолом и ацетатом натрия стандартными приемами, высушивается и повторно суспензируется в 15 мкл 10 Х буфера(50 ммоль Трис-, рН 7,5, 20 ммоль , 10 ммоль 2, 1 ммоль ДТТ), 5 мкл ограничительного фермента(20 ед.) и 130 мкл 2. После осторожного перемешивания реакционная смесь инкубируется при 37 С в течение ночи. ДНК снова осаждается этанолом и натрийацетатом, как указано выше, и подвергается электрофорезу на 15 -ном низкоплавком агарозном геле. Желаемый крупный ограничительный фрагмент срезается с геля, и ДНК извлекается путем осуществления процедуры в колонке -. ДНК выдерживается в 30 мкл 10 мМ Трис-, рН 8. Связывающие звенья ДНК, представленные на фиг. 31, химически синтезируются с помощью синтезатора(модели 380 В). После очистки каждого олигонуклеотида путем электрофореза на акриламидном геле они фосфорилируются, согласно описанной выше процедуре, и ренатурируются для ускорения сшивки и построения ДНК фрагментов, кодирующих аналог гена человеческого проинсулина. Связывающие звенья отдоиспользуются для построения плазмид 175, 176, 177 и 178 соответственно. Примерно 3 мкл фрагмента ДНК вектора 172, вываренного с /, смешиваются с 4 мкл каждого из киназированных связывающих звеньев (-) в буферном растворе (50 мкл), содержащем 50 мкмоль Трис-, рН 7,6, 10 ммоль 2, 10 ммоль ДТТ, 800 мкмоль АТР и 6 ед. Т 4 ДНК лигазы. Реакционная смесь инкубируется при 4 С в течение ночи, и эта смесь используется для трансформации клеток .К 12 308 согласно процедуре примера 29 3. Желаемые трансформанты .К 12 308/175, .К 12 308/176, . К 12 308/177 и .К 12 308/178 идентифицируются путем анализа их ДНК плазмиды и продуцирования белка до и после индукции клеток при 42 С. Ограничительная точка и функциональная карта плазмиды 175 представлены на фиг. 22. Аналогии человеческого проинсулина приведены в табл. 1. Е. Исходная культура .К 12 308/72 приготавливается в форме перманентной исходной культуры следующим образом. Выделенные колонии высеваются на нижеследующих фенотипных чашках -агар 5 мкг/мл Т (тетрациклина), -агар(стрептомицинсульфат), агаровая среда М-9 (солевая среда, содержащая 4, тиамин,АСЕ и глюкозу) и агаровая среда М-9, содержащая глюкозу и не содержащая лактозу. Одна чашка с -агаром/Тс инкубируется при 42 С, остальные чашки инкубируются при 30 С в течение 24 ч. Фенотипно 25 3225 1 правильная колония, отобранная с чашки -агар/Тс при 30 С, используется для инокулирования в колбе, содержащей 50 мл -питательного бульона (с содержанием триптона, дрожжевого экстракта,и глюкозы) плюс 5 мкг/мл среды Т. Эта колба инкубируется при 30 С со встряхиванием в течение 7 ч. Материал в колбе (1,2 мл) распределяется в бис-замораживающие ампулы и замораживается в парофазном жидком азоте. Затем материал оттаивает и используется в количестве 1 мл для инокуляции в колбе, содержащей 50 мл бульона плюс 5 мкг/мл Т. Эта колба инкубируется при 30 С со встряхиванием в течение 4 ч. Две колбы,содержащие 500 мл среды ВС-7 (содержит 4, тиамин, глюкозу, Т и изотопные соли), инокулируются с 1 мл каждой из указанных выше сред, и эти колбы инкубируются со взбалтыванием в течение 9 ч при 30 С. 150-литровый ферментер, содержащий 80 л среды (содержит лимонную кислоту, сульфат железааммония, К 2 НР 4, 41, 24, 2, 4, глюкозу и изотопные неорганические соли), инокулируется в 1 л питательного бульона ВС-7. Ферментер функционирует при 30 С до тех пор, пока скорость выделения двуокиси углерода не достигнет значения 1 ммоль/л/мин. В этот момент 50 л питательного бульона вводится в ферментер для получения клеточного инокулума. 2500-литровый ферментер, содержащий 1500 л той же среды, что используется в 150-литровом ферментере, функционирует при 30 С до тех пор, пока скорость выделения двуокиси углерода не достигнет значения 1 ммоль/л/мин, и затем температура повышается до 40 С. Функционирование ферментера продолжается еще в течение 8 ч. Затем питательный бульон инактивируется нагревом при 61 С в течение 7 мин. Из инактивированного нагреванием бульона получаются нижеследующие выводы выход клеток 13,1 г/л на сухой вес эффективность продукта 273 мкг -формилата/мл питательного бульона удельная активность (например, г продукта/г клеток) 2,1 процент формилированного продукта 13. Бульон брожения подается в сборник из нержавеющей стали, в котором температура поддерживается от 2 до 8 С. Затем весь питательный бульон центрифугируется или фильтруется для сбора клеток .так,чтобы выход твердых продуктов в расчете на сухой вес составлял 15-20 . Твердый продукт, составляющий собранные клетки, снова суспензируется в технологической воде. Затем клетки разрываются с использованием гомогенизации под высоким давлением или других подходящих методов так, чтобы лизис клеток составлял 90-99 . Гомогенизированная суспензия разбавляется до 5 сухой массы технологической водой. Величина рН разбавленной разорванной клеточной суспензии доводится до значения в пределах от 7 до 10 путем ввода гидрата окиси натрия. Инклюзионные образования (гранулы) отделяются от клеточных осколков путем дифференциального центрифугирования. Полученный концентрат содержит от 15 до 20 твердых веществ. Эти инклюзионные образования (гранулы) солюбилизируются при щелочном значении рН в присутствии цистеина и мочевины. Эти солюбилизированные гранулы подвергаются катионообменной хроматографии на текучей сефарозедля отделения смешанной дисульфидной формы проинсулинового аналога от общего объема гранулированного материала. Метионил-тиразоновое удлинение аминового конца аналога проинсулина удаляется путем инкубирования с катепсином С дипентидиламинопептидазой. Реакция катепсинового расщепления завершается путем сульфитолиза тетратионатом калия и цистеином при рН 8,8. После вытеснения растворителем черезС-25 полученный -сульфонат проинсулинового аналога подвергается перегибу путем инкубации при величине рН, равной 10,6, в присутствии цистеина. Процесс перегиба подавляется путем доведения величины рН до 2,4. Проинсулиновый аналог с должным перегибом отделяется от материала с неправильным перегибом а также от загрязнений, оставшихся после этапа расщепления катепсина С, путем хроматографического разделения с гидрофобным взаимодействием на смоле 20. Это разделение происходит с использованием ацетонового градиента, так что органический растворитель должен быть удален из основного потока, путем выпаривания перед дальнейшей операцией. После выпаривания органического растворителя проинсулиновый агент с правильным перегибом инкубируется с трипсином и карбоксипептидазой В. Эти ферменты эксцизируют С-пептид аналога проинсулина и образуют человеческий инсулин(В 28),(В 29). Этот аналог инсулина затем дополнительно очищается путем катионообменной хроматографии на -сефарозе с последующей хроматографией высокого разрешения с обратимой фазой на смоле С-8 (10 мкм). Ацетонитрил удаляется из основного потока обратимой фазы путем вытеснения растворителем наС-25, и полученный продукт концентрируется с использованием спирально намотанной системы ультрафильтрации. Концентрированный продукт подвергается хроматографии исключения на смолеС-50 и лиофилизируется до сухого состояния. В табл. 2 представлены данные аминокислотного анализа соединения, отвечающие изложенным выше примерам. Физиологические эффекты аналогов инсулина, показаны в нижеследующей испытательной системе в условиях. В качестве испытательных животных используются обычные самцы крыс, которые получены из. Полученные крысы имеют вес в диапазоне от 160 до 180 г, и их выдерживают в течение 1 нед в помещении при 75(24 С) с регулярным световым циклом (с включенным светом с 26 3225 1 7 вечера до 7 утра и с выключенным светом с 7 утра до 7 вечера). Рацион крыс составляет капуста Пурина 5001. Крысы, используемые для каждого испытания, подвергаются голоданию в течение примерно 16 дн до их использования. При первом использовании их вес составляет примерно 200 г. При достижении веса примерно 275 г (в период за 3 нед) животных уже больше не используют. Одна группа из 10 крыссамцов используется ежедневно для каждого испытываемого соединения (то есть биосинтезированный человеческий инсулин, свиной инсулин и аналог человеческого инсулина). В течение недели каждая группа используется лишь однократно. Десять крыс были разделены на две группы по пять крыс в каждой. Одна группа служит в качестве контрольной, и в нее вводится лишь носитель путем подкожной инъекции. Другая группа, состоящая из пяти крыс, подвергается обработке испытываемым соединением. Белки растворяются в 0,05 н. НС 1 (рН 1,6), и в результате получается исходный раствор концентрацией 100 мкг на мл. Из него получают ряд разбавленных растворов в обычном солевом растворе, который вводится путем подкожной инъекции в организм крыс. 100 мкл образца крови берут их хвостовой вены у каждой крысы при нулевом моменте времени и через 30 мин, 1 ч, 2 ч, 3 ч и 4 ч после еды. Глюкозу определяют колориметрическим способом оксидазы глюкозы (Со). Для каждой крысы рассчитывают изменение содержания глюкозы в крови от нулевого момента времени, и конечные результаты выражены как среднее изменение процентадля экспериментальной группы с корректировкой на среднее изменение для контрольной группы для того же дня. Кривая доза - реакция была построена по данным испытания с 4-7 различными концентрациями испытываемого соединения с использованием максимальной реакции в заданный момент времени (обычно 1 или 2 ч после ввода белка). Из данной кривой определяют значение 50, как значение вводимой подкожной дозы белка, которая дает половину максимальной гипогликемической реакции. Результаты представлены в табл. 3. Как указывалось выше, аналоги инсулина, имеют пониженную способность к димеризации или, иными остовами, способность к самоассоциированию высокомолекулярных форм. Так, при вводе одного или нескольких указанных аналогов достигается очень быстрое инициирование активности. Аналоги инсулина эффективны при лечении гипергликемии при вводе в организм пациента, нуждающегося в этом лечении, эффективного количества аналога инсулина формулы . Под используемым термином эффективное количество имеется в виду один или несколько аналогов инсулина, отвечающих данному изобретению, которые необходимы для снижения или поддержания уровня сахара в крови, либо терапевтически, либо профилактически. Это количество обычно может находиться в пределах примерно от 10 до 60 ед. -или более в день (или примерно от 0,3 до 2 мг, что составляет примерно 29 ед. на миллиграмм). Однако следует иметь в виду, что количество аналога (аналогов) инсулина, которое фактически вводится в организм, должно определяться лечащим врачом в соответствии с обстоятельствами, включающими состояние заболевания (то есть причину, вызвавшую гипергликемию), тип используемого аналога инсулина, который выбран для ввода в организм, выбранный парентеральный способ ввода, возраст пациента, вес и реакцию каждого пациента и остроту симптомов пациента. Следовательно, указанные выше пределы дозы никак не могут являться ограничением объема данного изобретения. Аналоги инсулина вводятся в организм пациента, нуждающегося в этом лечении (то есть пациента,страдающего от гипергликемии), с использованием фармацевтических композиций, содержащих эффективное количество по меньшей мере одного аналога инсулина формулыв композиции с одним или несколькими фармацевтически пригодными эксципиентами или носителями. Для этих целей фармацевтические композиции могут быть приготовлены таким образом, что будут содержать примерно 100 ед. на мл или аналогичные концентрации с содержанием эффективного количества аналога (аналогов) инсулина. Эти композиции обычно (хотя и необязательно) парентеральны по своей природе и могут быть приготовлены любым способом с применением обычных эксципиентов или носителей для вводимых парентерально продуктов, которые уже известны (см., например, публикацию, 17 е изд.,,, , 1985). Так, например, дозированные формы для парентерального ввода могут быть приготовлены путем суспензирования или растворения желаемого количества по меньшей мере одного аналога инсулина формулыв нетоксичном жидком носителе, пригодном для инъекции, таком как водная среда и стерилизующая суспензия или раствор. Как возможный вариант,измеренное количество соединения может быть помещено в ампулу, и ампула и ее содержимое стерилизуются и запаиваются. Ампула или носитель могут быть подготовлены для целей смешивания до ввода в организм. В фармацевтических композициях, пригодных для парентерального ввода в организм, используются разбавители, эксципиенты и носители, такие как вода и смешиваемые с водой органические растворители, такие как глицерин, сезамовое масло, арахисовое масло, водный пропилен-гликоль, .диметилформамид и т.д. Примерами таких фармацевтических композиций являются стерильные изотонические водно-солевые растворы аналогов инсулина формулы (1), которые могут быть приготовлены в форме буферного раствора с фармацевтически пригодным буфером и которые свободны от пирогенных веществ. Кроме того, вводимая парентерально фармацевтическая композиция может содержать предохра 27 3225 1 няющие средства, такие как мета-крезол, и другие вещества для регулирования величины рН конечного продукта, такие как гидрат окиси натрия или соляная кислота. Аналоги инсулина могут быть приготовлены также в форме фармацевтических композиций, пригодных для ввода через нос. Такие композиции описываются подробно в Европейской патентной заявке 2200383. Такие композиции приготавливаются вместе с одним или несколькими фармацевтически пригодными разбавителями,фармацевтически приемлемым количеством соли щелочного металла, соли аммония или свободной кислоты свободного от цинка инсулина, и при желании усиливающим абсорбцию количеством по меньшей мере одного усиливающего абсорбцию агента, выбранного из группы, включающей олеиновую кислоту или ее сложный эфир, жидкую форму сложного эфира сорбита и жирной кислоты, жидкую форму полиоксиэтиленового производного сложного эфира сорбита и жирной кислоты и жидкую форму сополимера оксиполиокси-этиленполиоксипропилен-полиоксиэтилен. Для демонстрации эффективности внутриносового ввода аналога человеческого инсулина(В 28), (В 29) в сопоставлении с человеческим натриевым инсулином приводится нижеследующее исследование. Самцы коротконогих гончих собак весом от 10 до 12 кг поддерживаются в прекрасном физическом состоянии до и в ходе данного исследования. Собаки выдерживаются в режиме голодания в течение 16 ч, но они имеют доступ к воде для гарантии предотвращения непредвиденных колебаний содержания инсулина. В течение всего исследования собак анестезируют пентабарбитолом натрия, и температура их тела сохраняется посредством нагревательных подушек для сведения к минимуму колебания содержания глюкозы. Испытательные образцы человеческого инсулина (то есть(В 28),(В 29) и человеческого натрийинсулина) растворяются в дистиллированной воде, и величина рН раствора доводится до 7,5 посредством гидрата окиси натрия. Конечная концентрация инсулина составляет 70 ед. на мл. Человеческий инсулин(В 28),(В 29) дозой 0,8 ед. на кг вводится в каждую из восьми собак, и человеческий натрийинсулин вводится дозой 0,8 ед. на кг в каждую из четырех собак. Все дозы вводятся через нос путем ввода струи через ноздрю с помощью дозировочного распылителя и модифицированного носового аппликатора. Образцы крови отбираются из шейной вены за 30, 15 и 0 мин до ввода и через 10, 20, 30, 45, 60, 90, 120, 180 и 240 мин после ввода для измерения снижения глюкозы в крови. Результаты данного исследования приводятся в табл. 4. При осуществлении описанной выше методики сопоставляется профиль снижения глюкозы крови при внутриносовом вводе человеческого инсулина(В 28),(В 29) с профилем снижения при внутривенном и подкожном вводе аналога. Для внутривенного ввода раствор инсулина, содержащий аналог человеческого инсулина(В 28),(В 29), вводится путем инъекции ударной дозы (0,1 ед. на кг) в подкожную вену ног каждой из четырех собак. Образцы крови берутся за 30, 15 и 0 мин до ввода и через 5, 10, 15, 20, 30, 45, 60, 90, 120, 180 и 240 мин после ввода. Для подкожного ввода раствор инсулина, содержащий аналог человеческого инсулина(В 28),(В 29), вводится под эпидермис (0,2 ед. на кг) другой боковой стороны живота каждой из шести собак. Образцы крови берутся таким же образом, как в случае носового ввода, как описано выше. Результаты данного сопоставительного исследования приводятся в табл. 5. Таблица 1 Аналог Плазмидабелка Человеческий проинсулин Аминокислотный анализ аналогов человеческого инсулина 1,2 3 4 5 6 7 8 4,00(4) 3,00(3) 2,81(3) 2,71(3) 2,71(3) 2,74(3) 7,18(7) 6,99(7) 1,02(1) 1,09(1) 4,01(4) 3,95(4) Продолжение таблицы 2 Аминокислотный анализ аналогов человеческого инсулина 1,2 3 4 5 6 7 8 9 1,04(1) 1,93(2) 1,02(1) 1,05(1) 1,05(1) 1,03(1) 1,03(1) Аминокислотный анализ аналогов человеческого инсулина 1,2 12 13 14 15 16 3,00(3) 3,00(3) 3,00(3) 3,00(3) 3,00(3) 3,59(3) 2,80(3) 2,80(3) 2,70(3) 2,83(3) 2,64(3) 2,43(3) 2,78(3) 2,87(3) 2,78(3) 6,84(7) 6,89(7) 7,09(7) 7,04(7) 7,08(7) 1,48(1) 1,09(1) 1,12(1) 0,93(1) 1,16(1) 5,70(5) 3,89(4) 4,03(4) 3,98(4) 4,00(4) 1,14(1) 1,04(1) 1,05(1) 1,08(1) 1,04(1) 4,54(6) 4,57(6) 5,19(6) 5,00(6) 4,75(6) 3,52(4) 3,58(4) 3,43(4) 3,41(4) 3,51(4) 1,82(2) 1,70(2) 2,36(3) 1,44(2) 1,51(2) 5,79(6) 5,95(6) 5,98(6) 6,89(7) 6,10(6) 3,89(4) 3,75(4) 3,74(4) 3,64(4) 3,86(4) 3,47(3) 3,01(3) 2,85(3) 2,71(3) 2,86(3) 2,24(2) 3,39(3) 2,07(2) 2,30(2) 2,48(2) 0,80(1) Аминокислотный анализ аналогов человеческого инсулина 1,2 21 22 23 24 25 3,00(3) 3,00(3) 3,00(3) 3,00(3) 3,00(3) 3,04(3) 2,76(3) 3,73(4) 2,70(3) 2,80(3) 2,69(3) 3,60(4) 2,74(3) 2,86(3) 2,78(3) 7,06(7) 6,99(7) 7,07(7) 7,01(7) 7,04(7) 2,20(2) 0,95(1) 1,00(1) 0,95(1) 1,11(1) 3,93(4) 3,91(4) 3,97(4) 3,94(4) 3,97(4) 1,02(1) 1,07(1) 1,03(1) 1,08(1) 1,04(1) 5,57(6) 4,31(6) 5,01(6) 4,05(6) 5,17(6) 3,61(4) 3,37(4) 3,42(4) 3,41(4) 3,43(4) Аминокислотный анализ аналогов человеческого инсулина 1,2 21 22 23 24 25 1,56(2) 1,42(2) 1,48(2) 1,44(2) 1,48(2) 6,09(6) 5,95(6) 6,02(6) 6,01(6) 5,98(6) 3,93(4) 3,60(4) 3,72(4) 3,78(4) 4,67(5) 3,13(3) 2,76(3) 2,85(3) 2,74(3) 2,83(3) 2,10(2) 2,55(2) 2,32(2) 2382(2) 2,08(2)(1) Молярная единица на основе аспарагиновой кислоты.(3) Половина цистина плюс аминомасляная кислота суммарно 6,49. Таблица 3 а) Максимальный эфБиологическая актив 50) Соединение фект,ность )(28), Ро(В 29)Н 1 53 8.7 91 а) Все показанные значения относятся к образцам крови, взятым через час после ввода указанного соединения.
МПК / Метки
МПК: A61K 38/28, C07K 14/62
Код ссылки
<a href="https://by.patents.su/30-3225-analog-insulina.html" rel="bookmark" title="База патентов Беларуси">Аналог инсулина</a>
Аналог витамина D

Номер патента: 1307
Опубликовано: 16.09.1996
Авторы: Мартин Йон КАЛВЕРЛЕЙ, Кай ХАНСЕН, Лизе БИНДЕРУП
МПК: A61K 31/59, C07C 401/00
Текст:
...используются для лечения человеческих болезней и в ветеринарии, как описано выше.Требуемое кошитчество соединения формулы 1 (в дальнейшем ссылаются на активный ингредиент) для достижения терапевтического эффекта, конечно, изменяется для данного Соединения в зависимости от пути приема и питания при лечетши. Соединения данного изобретения могут приниматься парентерально, внутрисуставно, ентерально или местным введением. Они хорошо...
Предыдущий патент: Трехпродуктовый гидроциклон
Следующий патент: Автоматизированная система охранной сигнализации “Алеся”
Случайный патент: Сбитень картофельный